Синдромом Прадера-Вилли в медицине называют редкое наследственное заболевание, для которого характерно отсутствие или недостаточное функционирование некоторых генов или их частей 15-ой отцовской хромосомы. Впервые данная патология была описана в 1956 году в Швейцарии педиатрами А. Прадером и Х. Вилли, по фамилиям которых и назван синдром. Частота его составляет 1 случай на 12-15 000 новорожденных детей. Симптомы и проявления синдрома Прадера-Вилли бывают различными, а течение заболевания зависит, как правило, от конкретного случая.
Общие сведения
Впервые синдром Прадера-Вилли упоминается Лэнгдоном Дауном в 1887 г. при описании 14-летней пациентки, страдающей задержкой роста, ожирением, снижением умственной деятельности и функции яичников (гипогонадизмом). Лэнгдом Даун назвал это заболевание полисарцией.
Классическое описание синдрома появилось в 1956 г. благодаря исследованиям пациентов с подобным фенотипом, проведенным швейцарскими педиатрами Андреа Прадером, Гвидо Фанкони, Генрихом Вилли и терапевтом Алексисом Лабхартом.
В 1965 г. был выявлен синдром Ангельмана, при котором мутации в этом же генетическом регионе (15q11-13) затрагивают не отцовскую, а материнскую копию хромосомы.
В 1981 г. при изучении 15-й хромосомы Ледбеттер и соавторы определили регион хромосомы, который при мутациях является причиной синдрома Прадера – Вилли.
Частота распространения заболевания колеблется от 1 к 10 000 до 1 к 25 000 новорожденных.
Заболевание одинаково часто наблюдается у мальчиков и девочек, и является основной генетически обусловленной причиной ожирения у лиц старше 1 года. Национальность и раса на распространенность заболевания не влияют.
Профилактика
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.
К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Формы
В 2000 г. на основе патогенеза исследователи (Олаецер и др.) выделили три фенотипа синдрома Прадера – Вилли:
- типичный фенотип синдрома Прадера- Вилли, развивающийся при делеции отцовской копии хромосомы;
- более легкий фенотип, отличающийся лучше развитой познавательной функцией (развивается при унипарентной материнской дисомии);
- выраженный фенотип с большим количеством сердечных патологий, развивающийся при материнской унипарентной дисомии и мозаичной 15 трисомии.
Примечания
- Buiting K. et al.
Clinical utility gene card for: Prader-Willi Syndrome (англ.) // European Journal of Human Genetics. — 2014. — P. e1–e3. — DOI:10.1038/ejhg.2014.66. - Prader A., Labhart A., Willi H.
Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter (нем.) // Schweiz. Med. Wschr. — 1956. — Bd. 1986. — S. 1260-1261. - Swaab, D.F. et al. (1995) Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J. Clin. Endocrinol. Metab. 80, 573—579
Причины развития
Синдром Прадера – Вилли возникает в результате нарушений функции участка q11—13 15-й пары хромосом (участок PWS-AS, ассоциированный с синдромами Прадера – Вилли и Ангельмана).
Развитие синдрома Прадера – Вилли всегда связано с хромосомой отцовского происхождения.
Заболевание развивается при:
- Делеции (утрате) региона q11—13 отцовской гаметы (выявляется в 70 % случаев).
- Полном отсутствии отцовской копии 15-й хромосомы и ее замене дуплицированной материнской копией (материнская дисомия). Наблюдается у 20 % пациентов.
- Функциональной дезактивации у плода в результате метилирования (модификации молекулы без изменения нуклеотидной последовательности ДНК) структурно нормального участка q11—13 отцовской хромосомы. Встречается у 5 % больных.
Авторы первого описания синдрома на основе проведенных наблюдений предполагали, что заболевание носит аутосомно-рецессивный тип наследования. Впоследствии наблюдавшиеся семейные случаи патологии дали основание предполагать аутосомно-доминантный тип наследования, однако большинство описанных случаев носило спорадический характер.
Дальнейшие исследования позволили установить, что синдром Прадера – Вилли у детей связан либо с транслокациями (хромосомными перестройками, при которых разные хромосомы обмениваются своими фрагментами), либо с мозаицизмом (наличии в тканях клеток, которые различаются генетически).
Наличие микроделеции 15 хромосомы при синдроме Прадера – Вилли было установлено в 1987 г., а молекулярно-генетические методы исследования позволили выявить роль геномного импринтинга и унипарентальной дисомии в развитии заболевания.
Долгое время считалось, что экспрессия (преобразование наследственной информации в белок или РНК) генов матери и отца равноценна, однако выявленный феномен геномного импринтинга продемонстрировал наличие избирательной экспрессии некоторых локусов хромосом в зависимости от их отцовского или материнского происхождения.
Точная причина унипарентальной дисомии в настоящее время не установлена, но обнаружено, что наследование двух хромосом только одного родителя является результатом серии генетических и биохимических нарушений (долгое время подобное наследование считалось невозможным, но при помощи молекулярно-генетических маркеров была установлена возможность такого наследования).
Диагностические мероприятия
Ранняя диагностика синдрома Прадера-Вилли и последующее лечение позволяют улучшить прогноз развития заболевания. Диагноз ставится, как правило, на основе клинических проявлений болезни, но на сегодняшний день часто используется генетическое тестирование, которое специалисты рекомендуют в первую очередь для новорожденных. Это обусловлено тем фактом, что у детей наличие синдрома определить намного труднее, поскольку невозможно проверить их способности, позволяющие проводить диагностику синдрома Прадера-Вилли по клиническим проявлениям. Генетическое тестирование проводится методом ДНК-метилирования с целью выяснения, присутствуют ли на 15 хромосоме отклонения, приводящие к возникновению заболевания. Данный способ диагностики синдрома Прадера-Вилли помогает выявить 97% случаев болезни. Стоит также отметить, что часто заболевание диагностируется неправильно, поскольку его нередко путают с синдромом Дауна, который встречается гораздо чаще. К тому же такой характерный признак синдрома Прадера-Вилли, как ожирение, может присутствовать также при синдроме Дауна. По этой причине огромное число случаев болезни остаются не выявленными.
Патогенез
Патогенез заболевания до настоящего времени остается малоизученным.
Существуют предположения, что ожирение, сопровождающее синдром Прадера – Вилли, обусловлено очень низкими процессами расщепления жиров (липолиза) и усиленным более чем в 10 раз синтезом жира из ацетата.
Гипогонадизм, развивающийся по гипогонадотропному типу, связывают с дисфункцией гипоталамуса (предположительно, патология затрагивает области вентромедиального и вентролатерального ядер). Данную теорию подтверждает эффективность лечения больных кломифеном, способствующим секреции гонадотропинов.
Свойственная больным гипопигментация кожи, волос и радужки объясняется снижением активности в волосяных фолликулах и в клетках, вырабатывающих меланин, тирозиназы – фермента, содержащего медь и катализирующего окисление фенолов.
Для 2-й фазы заболевания характерен дефицит СТГ (гормона роста), вызванный дисфункцией гипоталамуса.
Синдром Прадера – Вилли возникает благодаря импринтингу. Происходящая благодаря данному процессу детерминация экспрессии аллелей связана с их происхождением (всегда экспрессируется либо геном отца, либо геном матери). В результате отсутствия экспрессии альтернативной формы гена (отцовской) в фенотипе ребенка экспрессируется патогенная мутантная аллель матери.
Начало процесса импринтинга связано с процессом формирования гамет. В потенциально неактивной аллели генома эмбриона происходит метилирование цитозина и в ДНК образуются CpG-островки, которые состоят из пар гуанина (G) и метилированного цитозина (С). Благодаря метилированию в области промотора репрессируемого гена образуется абортивная (короткая, возникшая при преждевременном прекращении синтеза) РНК, необходимая для супрессии гена. Таким образом, посредством метилирования маркируется ген для импринтинга.
На возникновение однородительской дисомии оказывает влияние возраст матери (влияет на зачатия с трисомией или моносомией 15 хромосомы). Такие случаи объясняются материнским нерасхождением, при котором гомологичные хромосомы пары не расходятся, а направляются к одному полюсу клетки. После оплодотворения такой гаметы образуется зигота, содержащая нечетное число хромосом.
Лишняя хромосома до начала развития эмбриона во многих случаях подвергается удалению. Поскольку механизм удаления лишней хромосомы не учитывает ее происхождение, только в 66% случаев клетка избавляется от одной из материнских хромосом, а в остальных удаляется единственная отцовская.
Синдром Прадера – Вилли в 10 % случаев возникает в результате хромосомной перестройки (транслокации) в области критического для синдрома участка. Наблюдения за семьями больных с подобными транслокациями показали высокий риск рецидива (около 50 %) и зависимость фенотипа от пола передающего хромосомы родителя.
Фенотип синдрома Прадера – Вилли в 2 – 5 % случаев возникает при дефектах импринтинга. Дефекты, включающие разделение на 2 части центра импринтинга в пределах 15-й хромосомы (регион q11-q13), приводят к изменению эпигенотипа хромосомы одного из родителей. Данные дефекты часто связаны с делецией (утратой) центра импринтинга, а в некоторых случаях – с эпигенетической мутацией, которая не вызывает изменения последовательности нуклеотидов ДНК.
Во всех случаях выявленные дефекты приводят к изменениям в метилировании ДНК, нарушениям структуры хроматина и содержанию транскриптов данного гена в различных клетках.
Делеции в центре импринтинга связаны с 50%-ным риском рецидива (риск зависит от пола передающего аномалию родителя), а при отсутствии делеции риск рецидива невысок.
В настоящее время гены, которые затрагиваются геномным импринтингом в 15-й хромосоме (регион q11 -q13), выявлены лишь для синдрома Ангельмана. По предположениям ученых синдром Прадера – Вилли связан с генами в кластере некодирующих малых ядрышковых РНК ( snoRNAs ) либо с белок-кодирующими генами SNURF – SNRPN (расположен в области центра импринтинга) и Necdin.
Эндокринные нарушения
Существует несколько факторов, подтверждающих концепцию дефицита роста у лиц подверженных синдрому.
- Лица, подверженные болезни, имеют низкий рост и страдают от чрезмерного ожирения, то есть у них пониженное содержание свободной жировой массы, пониженная плотность костной ткани, низкий уровень использованной энергии.
- Для данной болезни характерны нарушения в мочеполовой системе. У мужского пола проблемы возникают с неопущением яичек (со временем яички могут опуститься до нормального уровня), а у женского в появлении адренархе. В обоих случаях возможно решение проблемы хирургическим путем.
Симптомы
На первом году жизни выявление синдрома Прадера – Вилли затруднено в результате отсутствия специфических симптомов при рождении и спонтанной ликвидации на протяжении первых месяцев жизни ранних признаков заболевания.
У детей с синдромом Прадера – Вилли часто наблюдается незначительная внутриутробная гипотрофия и асфиксия, возможна дисплазия тазобедренных суставов. Дети обычно рождаются доношенными. У 10-40% детей наблюдается ягодичное предлежание.
Ранние проявления синдрома включают:
- слабые шевеления плода;
- сниженный сосательный рефлекс после рождения (в некоторых случаях наблюдается его отсутствие);
- мышечную гипотонию, развивающуюся в первые месяцы жизни.
При нарушении акта сосания отсутствие зондового питания может в краткие сроки привести к развитию у ребенка дистрофии и других дефицитных состояний.
Мышечная гипотония приводит к нарушению моторного развития.
На протяжении первых месяцев жизни наблюдается спонтанное повышение тонуса мышц и восстановление сосательного рефлекса.
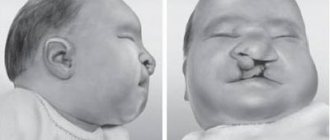
Уже на первом году жизни возможно проявление различных дисморфий лица и конечностей (акромикрия, для которой характерно наличие непропорционально маленьких стоп и кистей).
Дисморфия может проявляться наличием:
- удлиненной формы головы или микроцефалией;
- миндалевидного разреза глаз;
- широкой переносицы;
- косоглазия (страбизм);
- эктропиона (выворот века);
- тонкой верхней губы и маленького рта;
- низко расположенных ушных раковин и гипоплазии хрящей уха.
Также наблюдается гипогонадизм (для мальчиков характерен крипторхизм, гипоплазия мошонки и полового члена, а у девочек наблюдается недоразвитие половых губ и в половине случаев матки).
Дети с синдромом Прадера – Вилли отличаются повышенной сонливостью, наличием густой слюны и проблемами с зубами (микродонтия и др.). В 75% случаев от общего числа заболевания наблюдается слабая пигментация волос, радужки и кожи.
Также часто выявляется сколиоз и пониженная плотность костей.
Синдром Прадера – Вилли обычно диагностируют при развитии второй фазы заболевания, для которой характерно:
- Появление ощущения постоянного чувства голода и формирование поведения «постоянного поиска пищи» (полифагия).
- Быстрое развитие ожирения (жировые отложения наблюдаются преимущественно на теле и в проксимальных отделах конечностей).
- Замедление процессов роста. Без лечения окончательный рост мальчиков составляет 155 см, а девочек – 147 см.
- Проявление умеренных признаков нарушения интеллекта (при норме коэффициента интеллектуального развития 85-115 ед. у больных наблюдается от 20 до 80 ед.). Словарный запас ниже нормы, речь больных обычно затруднена.
- Нарушение сроков полового развития и изменение порядка появления вторичных половых признаков.
Настроение у больных с симптомом Прадера – Вилли характеризуется частой сменой, но в целом они доброжелательны.
Встречаются в отдельных случаях судороги и нарушения координации.
Присутствующая во многих случаях дневная сонливость протекает у некоторых больных по типу нарколепсии, наблюдаются эпизоды ночных апноэ.
Возможно развитие сахарного диабета с тенденцией к возрастному улучшению.
ЯМР-томография в 12% случаев выявляет аномалии коры головного мозга и кисты червя мозжечка.
Симптоматика
Симптомы у синдрома Прадера-Вилли различные, меняются в зависимости от возрастной категории больного.
Внутриутробно патологическое состояние имеет такие признаки:
- пониженная активность движений плода;
- необычное расположение;
- многоводие.
Сразу после рождения симптоматика следующая:
- ягодичное предлежание плода;
- пониженное давление;
- снижение сосательного рефлекса;
- затрудненное дыхание.
В детстве у ребенка отмечают такие состояния:
- заторможенное развитие навыка речи;
- поражение зубов кариесом;
- угнетенная координация движений;
- употребление большого количества пищи;
- быстрый набор лишнего веса;
- проблемы со сном;
- сколиоз;
- половое созревание наступает позже обычного;
- невысокий рост;
- задержка интеллектуального развития;
- задержка психомоторного развития;
- чрезмерная гибкость.
Признаки в совершеннолетнем возрасте:
- бесплодие;
- небольшое количество волос в области лобка;
- ожирение;
- склонность к сахарному диабету.
Обобщенная симптоматика внешних отличий у взрослых людей включает:
- нос больших размеров, широкий;
- узкие пальцы на руках;
- конечности маленького размера;
- присутствует гиперчувствительность кожного покрова;
- большое количество избыточного веса;
- лоб высокий и узкий;
- кожный покров и волосы более светлого оттенка по сравнению с родственниками.
Наблюдается задержка моторного и полового развития.
Диагностика
Поскольку выявление симптома Прадера – Вилли до 2-й фазы заболевания позволяет сформировать у ребенка правильное пищевое поведение, а начатая до 18-месячного возраста коррекция дефицита гормона роста способствует формированию правильного телосложения, ранняя диагностика симптома очень важна.
Диагноз основывается на оценке имеющихся у больного признаков, которые подразделяются на большие и малые. Синдром Прадера – Вилли предполагается у детей до 3-х лет при наличии минимум 5 баллов, а после 3-х лет – при наличии 8 баллов, из которых 4 относятся к большим признакам заболевания.
Большие признаки, оценивающиеся в 1 балл, включают:
- Характерные черты лица. К таким чертам относят долихоцефалию с уменьшением диаметра в области височных костей, миндалевидный разрез глаз и страбизм, маленький рот, углы которого опущены вниз, тонкую верхнюю губу.
- Задержку нервно-психического развития в возрасте до 6 лет и умеренное снижение интеллекта.
- Проблемы с кормлением в первые месяцы жизни, сменяющиеся нормализацией сосания.
- Изменения в половой сфере.
- Мышечную гипотонию центрального генеза, которая присутствовала в раннем детстве.
- Прогрессирующее от 1 года до 6 лет ожирение.
Малые признаки (0,5 балла каждый) включают:
- сниженную двигательную активность плода и наличие инфантильной летаргии;
- нарушения рефракции;
- пониженную по сравнению с родителями пигментацию кожи и волос;
- «потертость» кожных покровов;
- наличие поперечной ладонной складки;
- низкорослость в возрасте 15 лет по сравнению с другими членами семьи;
- наличие нарушений сна и эпизоды апноэ;
- маленький размер стоп и кистей;
- наличие дефектов артикуляции и речи;
- вязкость слюны;
- нарушения поведения.
Несмотря на высокую чувствительность шкалы, постановка диагноза требует проведения кариотипирования (в большинстве случаев кариотип соответствует норме) и молекулярно-генетических исследований 15-й пары хромосом.
Для выявления микроделеции или унипарентальной дисомии применяют методы прометафазного анализа, использования ДНК-маркеров определенных участков хромосомы 15 и др.
Необходим дифференциальный диагноз с синдромом Лоуренса-Муна-Барде-Бидля, адипозогенитальной дистрофией, врожденной миопатией, врожденной спинальной амиотрофией и синдромом Альстрема.
Материал и методы
Все больные были направлены в лаборатории Научного центра психического здоровья РАМН и Московского НИИ педиатрии и детской хирургии для подтверждения диагноза.
Предварительный генетический анализ больных детей был проведен с применением различных цитогенетических и молекулярно-цитогенетических диагностических тестов на наиболее частые генные и хромосомные синдромы, ассоциированные с аутистическими расстройствами и синдромальными формами умственной отсталости, включая FISH для анализа случаев хромосомного мозаицизма и субтеломерных микроделеций и дупликаций [1, 4, 13, 15-22]. Молекулярное кариотипирование в этой когорте детей было описано нами ранее [5, 6, 10]. В данной работе проведены цитогеномные исследования 30 детей с предполагаемым диагнозом синдромов Ангельмана или Прадера-Вилли.
In silico
или биоинформатический анализ выявленных вариаций генома (CNV) проводили с использованием следующих баз данных: DECIPHER (database of unbalanced chromosome aberrations) — decipher.sanger.ac.uk, OMIM (online Mendelian inheritance in Man) — www.omim.org, The Phenotype-Genotype Integrator (PheGenI) — www.ncbi.nlm.nih.gov/gap/PheGenI, SFARI Gene/AutDB (web-based searchable database for autism research) — www.mindspec.org/autdb.html, Catalog of Published Genome-Wide Association Studies (NHGRI) www.genome.gov/gwastudies.
Лечение
Поскольку синдром Прадера – Вилли относится к врожденным генетическим аномалиям, специфические способы его лечения отсутствуют.
Терапия направлена на нормализацию обмена жиров и углеводов, а также на устранение симптомов заболевания.
При лечении больных используются:
- диетотерапия, включающая учет калорийности пищи и контроль за режимом питания;
- оральные гипогликемические препараты для лечения сахарного диабета;
- кломифен, восстанавливающий секрецию гонадотропинов и половых стероидов;
- гормон роста.
На раннем этапе заболевания при гипотонусе назначается массаж.
При выявлении нарушений биоэнергетического обмена назначаются витамины (Е, С, В2, тиамин, витамин РР), коэнзим Q10, улучшающий кровообращение циннаризин.
Медикаментозное лечение дополняется занятиями с логопедом и дефектологом.
Дети с синдромом Прадера – Вилли постоянно находятся под наблюдением эндокринолога, невролога, психотерапевта и офтальмолога.
Прогноз продолжительности жизни
Тяжесть течения болезни зависит от того, насколько удается компенсировать диетой и препаратами нарушения углеводного и жирового обмена. Сахарный диабет и ожирение приводят к тяжелым сосудистым осложнениям, инфаркту, инсульту. С ними связаны основные риски для жизни.
Способность ДНК к самовосстановлению снижена, в пожилом возрасте это приводит к повышенному риску опухолевых процессов. При хорошем контроле за состоянием здоровья пациенты доживают до 65-ти и более лет.