General information
Amyloidosis is a group of diseases characterized by the deposition of pathological insoluble proteins with a peculiar fibrillar structure in organs and tissues.
With a systemic disease, not only the process of protein metabolism is disrupted, but also the normal functioning of the immune system. The specific protein-saccharide complex amyloid can be deposited in all tissues of the body, gradually displacing healthy and normal cells from the affected organ. All this leads to functional dysfunction of the organ, and with a long course, multiple organ failure develops with a fatal outcome. Only 1% of the population suffers from this rare disease, but the incidence of certain forms of amyloidosis varies. The most common diagnosis is secondary amyloidosis. In the countries of the Mediterranean basin, genetically determined, hereditary amyloidosis predominates. A similar trend is observed among persons of Jewish and Armenian nationality.
Among the clinical forms, the most common is the nephropathic form with predominant damage to the renal system, as well as the generalized form, in which all organs and systems are affected. The incidence among men is 2 times higher than among women.
Some facts:
- In different forms of the disease, the chemical structure of amyloid is very different. Amyloidosis is a group of diseases that are similar in clinical symptoms, development mechanism and pathological manifestations, but the amyloid protein itself can be different.
- The disease may not manifest itself in any way for decades.
- There are no radical methods of therapy that can immediately get rid of the pathology.
- The true causes of the development of some forms of amyloidosis are still unknown to science.
Pathogenesis
Mechanism of amyloid formation:
- Formation of amyloid lasts. At the initial stage, mutational changes occur in various cells of the immune system. Monocytes, cells of the macrophage system and lymphocytes mutate, turning into completely new cells - amyloidoblasts. The number of amyloidoblasts increases as the disease progresses, gradually accumulating in certain organs (liver, spleen, lymph nodes, bone marrow).
- Synthesis of fibrillar protein. The amyloid scaffold is formed from long, thin filamentous structures, which are based on fibrillar protein formed within the amyloidoblast. For the body, fibrillar protein is foreign, so protective mechanisms are launched in response to their appearance. Amyloidoblasts begin to surround other cells, which destroy by absorption the fibrillar protein released from the amyloidoblasts. Such processes in the body can take place for an indefinitely long time in the form of a hidden, latent phase, but ultimately immunological tolerance will still develop, which will provoke an even greater flow of fibrillar protein into the tissue.
- Amyloid deposition in tissues. Amyloid is formed in the intercellular space after the fibrillar protein combines with nucleoproteins and proteins of tissues and blood. Such a bond is considered very strong and it is almost impossible to destroy it, which leads to the accumulation of amyloid in tissues, gradually displacing healthy, normal cells of a certain organ.
Cells that can develop into amyloidoblasts:
- cardiomyocytes ( cardiac amyloidosis );
- c-cells of the thyroid gland;
- beta cells of the pancreas that produce insulin (insular amyloidosis);
- keratinocytes ( cutaneous amyloidosis );
- smooth muscle ( large vessel amyloidosis );
- cells of the APUD system.
The above mechanism is not typical for all forms of amyloidosis, but for most. In certain cases, amyloid is formed by a different mechanism, which has not yet been fully studied.
Associated disorders
The following disorders may be associated with amyloidosis. Amyloidosis may occur in combination with or as a result of the following disorders:
Multiple myeloma, lymphoma, Hodgkin's lymphoma, medullary thyroid carcinoma, Whipple's disease, Crohn's disease, osteomyelitis, rheumatoid arthritis, ankylosing spondylitis, Reiter's syndrome, psoriatic arthritis, tuberculosis, macrodisease, congenital purulent-venous disease (congenital intestinal hyperplasia, hereditary congenital rihydrocytogenesis )
Classification
Modern classification is based on the specificity of the main protein that forms amyloid. Initially, the type of amyloid is indicated, then the precursor protein, and then the clinical form of the disease with a description of the affected target organs. The type of amyloid in all names begins with the letter “A”, which implies “amyloid”, then the specific fibrillar protein is indicated in the form of an abbreviation:
- AA amyloidosis. Here the second “A” denotes an acute-phase protein, which is produced in response to the presence of a neoplasm or an inflammatory process.
- AL amyloidosis. Here “L” stands for immunoglobulin .
- ATTR amyloidosis. Here “TTR” is the transport protein transthyretin for thyroxine and retinol .
- A-beta2-M amyloidosis. Beta-2-microglobulin is encrypted here .
AA amyloidosis. AA amyloid is formed from serum acute phase protein, which is a globulin that is synthesized by hepatocytes, neutrophils and fibroblasts. Its amount increases many times during inflammation or the presence of tumors. However, only certain fractions of amyloid participate in the formation of amyloid, so amyloidosis develops only in a portion of patients with inflammatory or tumor diseases. The final stage of amyloidogenesis—polymerization of the soluble precursor into fibrils—is not fully understood. It is believed that this process occurs on the surface of macrophages with the participation of membrane enzymes and tissue factors, which determines organ damage.
AA amyloidosis is characterized by 3 forms:
- Secondary reactive amyloidosis , which occurs against the background of tumor diseases and inflammatory processes. It is considered the most common form. Among the reasons for the development of the secondary form, the following came to the fore: rheumatoid arthritis ; diseases of the blood system ( lymphogranulomatosis , lymphoma ); psoriatic arthritis ; Crohn's disease , ulcerative colitis . The disease can develop against the background of osteomyelitis , tuberculosis and other purulent-obstructive pathology .
- Familial Mediterranean fever (periodic disease) with autosomal recessive inheritance. Ethnic predisposition is noted among Armenians, Arabs, Gypsies and Jews. There are 4 forms: articular; febrile; abdominal; thoracic The disease begins with unexplained fever and manifestations of arthritis . 40% of patients develop renal amyloidosis .
- Familial nephropathy with deafness and urticaria ( Muckle-Wales syndrome ). The disease is inherited in an autosomal dominant manner. a rash , urticaria , Quincke's edema appears , and hearing decreases. Finally, renal amyloidosis . This variant is considered the most common among inherited amyloidosis.
Target organs for AA amyloidosis: kidneys; liver; spleen; adrenal glands; intestines.
AL amyloidosis. The pathology is formed from the light immunoglobulin pathways, in which the sequence of amino acids is disrupted, which causes destabilization of the molecules and leads to the formation of amyloid fibrils. Local factors take part in the development of the pathological process, depending on which one or another organ is affected. The synthesis of immunoglobulins is carried out by an abnormal clone of beta and plasma cells in the bone marrow, which most likely resulted from a mutation or T-immunodeficiency.
There are 2 forms:
- idiopathic primary amyloidosis, which was not preceded by any disease;
- amyloidosis in beta cell tumors and myeloma.
Target organs for AL amyloidosis:
- digestive tract;
- heart;
- nervous system;
- kidneys;
- skin.
In AL amyloidosis, a deficiency of coagulation factor X leads to the development of a hemorrhagic syndrome with specific hemorrhages around the eyes, known as “raccoon eyes.”
In differential diagnosis, it is taken into account that the AA type is the youngest and affects people with an average age of 40 years, and for the AL form the average age is 65 years. In both cases, the patients are predominantly male.
ATTR amyloidosis includes 2 variants:
- Familial neuropathy (rarely nephropathy and cardiopathy). Inherited in an autosomal dominant manner. Amyloid is formed from mutant transthyretin, which is synthesized by liver cells and hepatocytes. Mutant proteins are characterized by instability and under certain conditions they transform into fibrillar structures, forming the amyloid protein.
- Senile familial amyloidosis. Occurs only in elderly people. The pathology is based on non-mutant transthyretin, normal in amino acid composition, whose physicochemical properties are changed. Fibrillar structures are formed under the influence of age-related metabolic changes in the body. The disease most often affects the nervous system, less often the heart and renal system.
A2M amyloidosis. A new manifestation of systemic amyloidosis, which appeared after the introduction of chronic hemodialysis into practice. The culprit protein is considered to be beta-2-microglobulin, which during hemodialysis cannot be filtered and is retained in the patient’s body. Amyloidosis develops after an average of 7 years of regular hemodialysis. The target organs are the periarticular tissues and the skeletal system.
Local amyloidosis. It can develop at any age and affect any organ or tissue. In practice, pancreatic islet amyloidosis is considered in elderly patients. Facts are described that confirm that almost all cases of diabetes mellitus in the elderly are in one way or another pathogenetically associated with amyloidosis of the pancreatic islets, which is formed from a beta-cell polypeptide.
Cerebral amyloidosis is considered the basis of Alzheimer's dementia . Whey protein is deposited in brain neurofibrils, atherosclerotic plaques, and vascular stacks.
Affected Populations
It is estimated that approximately 4000 new cases of AL amyloidosis are reported annually, although the actual incidence may be slightly higher as a result of underdiagnosis. Although the incidence is thought to be similar in men and women, about 60% of patients admitted to the centers are men. AL amyloidosis occurs in people as young as 20 years of age, but is usually diagnosed between the ages of 50 and 65 years.
People at risk of developing AA amyloidosis include people with chronic inflammatory diseases such as rheumatoid arthritis, psoriatic arthritis, chronic juvenile arthritis, childhood ankylosing spondylitis, and inflammatory bowel disease.
People with chronic infectious diseases such as tuberculosis, leprosy, bronchiectasis, chronic osteomyelitis and chronic pyelonephritis are also at risk. Secondary amyloidosis (AA) occurs in less than 5% of people with these conditions.
Causes
Amyloidosis can be an independent disease or develop against the background of another pathology of the body. For reasons of development, it is customary to distinguish:
- idiopathic (primary) amyloidosis;
- reactive (secondary) amyloidosis;
- amyloidosis in tumor neoplasms;
- senile amyloidosis;
- hereditary amyloidosis;
- amyloidosis in patients receiving hemodialysis.
Primary amyloidosis
In primary amyloidosis, the true cause of amyloid formation cannot be determined. In almost all cases, in the primary form of the disease, degeneration of immune system cells is recorded by a mutational mechanism with the formation of AL-amyloid, which accumulates mainly in tissues of mesodermal origin (nerves, cardiovascular system, skin, muscular system, etc.).
AL amyloid can also form in plasmacytoma ( myeloma ), a tumor pathology characterized by malignant degeneration and proliferation of differentiated B lymphocytes (plasmocytes). It is these cells that secrete abnormal globulins in huge quantities, which react with plasma nucleoproteins and form amyloid protein.
Secondary amyloidosis
This form is characterized by the formation of AA amyloid. The disease develops against the background of a long-term progressive inflammatory process in the body, being essentially a complication of another disease.
Causes of secondary amyloidosis:
- chronic purulent diseases (prolonged suppuration of wounds, osteomyelitis );
- chronic foci of infection ( syphilis , tuberculosis , leprosy , pyelonephritis , etc.);
- tumors, neoplasms ( leukemia , lymphogranulomatosis and other tumor lesions of the blood system);
- nonspecific ulcerative colitis (disease of the large intestine of inflammatory origin);
- rheumatological diseases ( ankylosing spondylitis , rheumatoid arthritis , etc.).
With secondary amyloidosis, absolutely any human organs and systems can be affected. Only a few years after the onset of the disease, pronounced clinical symptoms appear in the form of a functional disorder of the affected organ (spleen, liver, lymph nodes, adrenal glands, kidneys). Multiple organ failure develops gradually and can cause death.
Hereditary amyloidosis
Each of the 23 pairs of chromosomes that form the human genetic apparatus contains a huge number of genes. And each of the genes is responsible for the formation of certain specific proteins in the cell, which further determines its functionality and structure. Hereditary amyloidosis is caused by genetic mutations in the body's immune cells, which are passed on from generation to generation and are responsible for the formation of amyloidoblasts. The hereditary form is detected in individuals of certain ethnic groups or in people living in certain territories.
Diseases that belong to hereditary forms of amyloidosis:
- Muckle and Wells disease (English amyloidosis, familial nephropathic amyloidosis). Characterized by attacks of fever, changes in auditory perception (even deafness), and urticaria.
- Familial Mediterranean fever (periodic disease). Pathology is registered among Armenians, Jews and Arabs. Patients experience inflammation of the serous membranes, an increase in body temperature above 39 degrees, fever and mental abnormalities.
- Hereditary cardiopathic amyloidosis. Clinically very similar to generalized primary amyloidosis and occurs mainly in residents of Denmark.
- Hereditary neuropathic amyloidosis. The nervous system innervating the arms, legs, eyes, kidneys, etc. is affected.
Senile amyloidosis
Individuals over 80 years of age may experience characteristic deposition of amyloid protein in various tissues and organs. The pathological process is most often associated with other diseases and is local in nature. Forms of senile amyloidosis:
- Cerebral amyloidosis (senile, cerebral). It is characterized by the deposition of Ab type amyloid protein in brain tissue and occurs in Alzheimer's disease.
- Senile cardiac amyloidosis . It affects the heart muscle, and the culprit may be different types of amyloid (some are deposited in the atria, and some in the ventricles). In both cases, amyloid can accumulate in parallel in other organs (pulmonary system, spleen, pancreas).
Amyloidosis in tumors
Some tumors are characterized by degeneration of cells in the affected organ, which leads to the production of a specific fibrillar protein. In such cases, amyloidosis is local in nature. Causes of amyloidosis:
- Pancreatic islet tumor. The islets are a cluster of specific cells that produce various hormones ( glucagon , insulin , somatostatin , etc.). During malignant degeneration of such cells, fibrillar protein is produced, which later turns into amyloid.
- Medullary thyroid cancer. The neoplasm is formed from the C-cells of the thyroid gland, which normally produce the hormone calcitonin , which controls calcium metabolism in the body. In this case, AE-type amyloid is formed, which consists of calcitonin with altered synthesis.
Amyloidosis in hemodialysis patients
Hemodialysis refers to the artificial purification of human blood from metabolic by-products that are excreted through the renal system during its normal functioning. “Artificial kidney” is indicated for patients with acute and chronic renal failure. The essence of the procedure is the passage of blood through a special apparatus, which helps remove excess fluid and various harmful substances from the blood passing through. After purification, large proteins and blood cells are returned to the body.
During long-term hemodialysis, amyloid is formed from B2-microglobulin , which is normally removed exclusively through the renal system, but is not filtered through the dialyzer membrane, retaining and accumulating in the human body. Patients with renal failure are on hemodialysis for many months and years, which helps to increase the level of microglobulin-B2 several times. After binding to plasma nucleoproteins, the complex precipitates in tissues and organs (including the kidneys, aggravating the course of renal failure).
Causes of amyloidosis – who is at risk?
The following pathological conditions can provoke the disease in question:
- Multiple myeloma. In this pathology, plasma cells are modified and produce atypical globulins. Amyloid is formed due to the entry of degenerative globulins into a chemical reaction with plasma nucleoproteins.
- Oncopathology of blood.
- Infection of the respiratory system, kidneys, which becomes chronic: tuberculosis, pyelonephritis.
- Malaria.
- Rheumatological diseases.
- Chronic ailments that are characterized by the formation of purulent discharge : osteomyelitis, etc.
- Alzheimer's disease . In this case, amyloid is concentrated in the brain tissue.
- Acute or chronic renal failure , in which the patient remains on hemodialysis for several months/years.
- Malignant neoplasms , regardless of their location.
An important risk factor for the development of the disease in question is the natural aging of the body. As people age, they are susceptible to certain senile diseases, which can lead to the deposition of harmful substances in the brain tissues of the body, as well as in the heart.
Video: Renal amyloidosis and periodic disease
Symptoms of amyloidosis
Considering that amyloidosis is characterized by damage to any organ and tissue, the clinical picture can be very diverse. At the initial stages, the disease may not manifest itself in any way, but gradually disturbances in the functional state of a certain organ are registered, and organ failure develops.
As amyloidosis progresses, other organs and tissues become involved in the pathological process. Local amyloidosis, which is characterized by damage to only one organ, is extremely rarely recorded.
What is affected by amyloidosis:
- renal system;
- the cardiovascular system;
- adrenal glands;
- spleen;
- joints;
- muscle;
- digestive tract;
- hepatic system;
- skin;
- nervous system.
Kidney amyloidosis
Most often, amyloidosis affects the renal system, and kidney damage is considered the most dangerous manifestation of the disease. Symptoms of renal amyloidosis depend on the stage of the disease.
It is customary to distinguish 4 stages:
- Hidden (latent) stage . Clinically, the patient has no symptoms. In the secondary form, symptoms of the underlying disease predominate, and symptoms of renal amyloidosis appear after several years or decades. In medical practice, cases of rapid development of pathology are described - after 3-4 months.
- Proteinuric stage . Amyloidosis deposits are deposited in glomeruli, small vessels, and intercellular space. The stage may last 10 or more years. As the pathology progresses, compression of tissues occurs, their atrophy and subsequent death of nephrons. As a result of a violation of the filtration ability of the kidneys, protein begins to be excreted in the urine. At this stage, the excretory function of the kidneys is impaired, but continues to function, which makes diagnosing the disease difficult. The diagnosis can only be confirmed through laboratory testing.
- Nephrotic stage . It is characterized by further destruction of the renal filter, which leads to the formation of nephrotic syndrome with the release of large amounts of protein in the urine (more than 3 g/l) and a decrease in the concentration of proteins in the blood. Proteins take part in the retention of the liquid part of the blood - plasma in the vascular bed, maintaining colloid-osmotic pressure. Therefore, when they decrease, fluid retention in the body is observed and generalized edema , which makes it possible to suspect amyloidosis at this stage. Swelling is observed at any time of the day and does not depend on body position. First, the face and eyelids swell, then the swelling descends to the abdomen, lumbar region and genitals. As the pathology progresses anasarca , the skin becomes pale, and the legs and hair become dry and brittle. Blood may be found in the urine. The nephrotic stage can last 4-6 years, after which it passes into the next stage.
- Azotemic stage . The kidney system only does 25% of its work. There is a delay in metabolic products (uric acid, urea, etc.).
Clinical manifestations of renal failure:
- Changes in urine formation. Normally, a person should excrete 800 ml of urine, but with renal failure, the volume is reduced to 50 ml.
- Deterioration in general health. It manifests itself as excessively rapid fatigue and weakness.
- Changes in the functioning of the digestive tract. Vomiting, bad breath, dry mouth, and nausea appear.
- Changes on the skin. Patients complain of skin itching, pallor, scratching; changes in the cardiovascular system. Arrhythmia appears , blood pressure rises, and the cavities of the heart increase.
- Brain damage. High concentrations of uric acid and urea have a negative effect on brain structures, which is manifested by memory impairment, insomnia , irritability, changes in mental abilities and psyche.
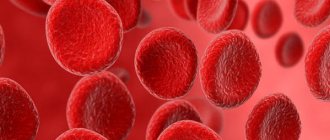
- Anemia. The number of red blood cells and hemoglobin concentration are reduced due to a lack of erythropoietin, a specific substance that is synthesized by the renal system and activates the formation of red blood cells in the bone marrow.
A microscopic specimen of a kidney affected by amyloidosis is stained with Congo stain. At low magnification, you can see protein deposits in the form of small blocks of various shapes, painted red or brick. The largest amount of amyloid is recorded in the glomerular apparatus, and amyloid is contained in smaller quantities outside the glomeruli and in the walls of the arteries.
Liver amyloidosis
Damage to the hepatic system is often recorded in the generalized form of amyloidosis. As a result of amyloid deposition, compression of liver cells, liver blood vessels and bile ducts occurs, impairing their function.
The deposition of amyloid deposits leads to an increase in the liver, which can be detected by palpation during the initial examination of the patient. The surface of the liver remains smooth, its lower edge is even, there is no pain. Liver failure develops extremely rarely , even with a long course of the process, which is associated with the high regenerative abilities of the liver.
Symptoms of liver amyloidosis:
- Increase in organ size - hepatomegaly . An enlarged and dense liver is detected by palpation.
- Signs of portal hypertension. Blood from the internal organs (spleen, intestines, stomach) enters the portal vein, after which it is sent to the liver, where the detoxification process takes place and then returns to the systemic circulation. As a result of compression of blood vessels by amyloid protein deposits, there is an increase in blood pressure in the portal vein system, which compensatory leads to expansion of the venous system of internal organs, up to their rupture. All this can lead to swelling of the lower extremities, ascites , bleeding from the digestive tract, and bowel dysfunction.
- Jaundice . Extremely rare with amyloid deposition. It develops against the background of impaired outflow of bile through the biliary tract due to compression by deposits from the outside.
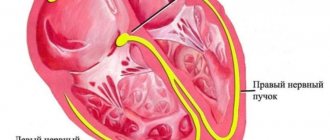
Cardiac amyloidosis
The myocardium is affected in some forms of hereditary amyloidosis and in the primary form. Amyloid protein can be deposited not only in the heart muscle, but also in the lining of the heart (endocardium, pericardium). Amyloid deposits compress the coronary arteries and negatively affect the blood circulation of the heart, which leads to direct death of muscle cells. Clinical manifestations:
- restrictive cardiomyopathy ;
- rhythm disturbances;
- heart failure.
Amyloidosis of the spleen
As a result of the deposition of deposits, the spleen increases in size, which makes the organ accessible for palpation (normally the spleen cannot be palpated). The surface of the organ is smooth and even, the structure is dense, painless. Symptoms of spleen amyloidosis:
- thrombocytopenia;
- anemia;
- leukopenia.
The micropreparation has a greasy sheen. The sebaceous (amyloid falls out diffusely) and sago (amyloid falls out in grains) spleen are distinguished. Usually the protein falls out along the reticular fibers.
Intestinal amyloidosis
Intestinal damage in the generalized form of amyloidosis is widespread, which negatively affects the absorption of vitamins , nutrients and minerals.
Symptoms of intestinal amyloidosis:
- frequent loose stools;
- muscle weakness;
- weight loss (up to cachexia);
- fast fatiguability;
- general weakness, lethargy, apathy.
- hair loss, brittle nails;
- anemia;
- mental, mental disorders;
- swelling (due to reduced protein content in the blood).
With local amyloidosis, protein can accumulate in one area, simulating a neoplasm or tumor that compresses the intestine, disrupting its peristalsis. First symptoms:
- pain in the epigastric region;
- bloating;
- constipation;
- vomiting of feces (in extremely severe cases).
Amyloidosis of the skin
Amyloid changes the structure of the skin and its blood supply, which manifests itself in the form of peeling, dryness and pallor. In some forms of pathology, a rash appears in the form of many dense, small blisters of red or yellow color. Possible fusion of bubbles, which leads to an increase in the affected area. Pain and itching are extremely rare. There may be peeling above the surface of the nodules. Photo of skin amyloidosis below.
Amyloidosis of the nervous system
When amyloid protein is deposited, impulse transmission along nerve fibers is disrupted, which manifests itself in certain symptoms depending on the affected area. Senile amyloidosis is characterized by damage to the tissues and structures of the brain, which is manifested by a decrease in mental abilities, intelligence, attention and memory. Possible mental disorders. In the familial form, the peripheral nervous system is affected, which is manifested by a crawling sensation, itching, soreness and impaired sensitivity in certain areas. In severe cases, paralysis - complete loss of motor activity.
Amyloid bodies in prostate secretions
In the analysis of prostate secretions, specific clots are identified. Amyloid bodies are oval in shape and should not normally be detected. Their presence indicates an inflammatory process. With congestion in the prostate gland, the number of amyloid bodies increases. Identification of pathological elements may indicate the development of prostate adenoma .
Most often, amyloid bodies are diagnosed in elderly people.
Standard therapy
The treatment strategy depends on the type of amyloidosis and the clinical condition of the patient. In AL amyloidosis, the cause is an abnormal white blood cell (usually a plasma cell), and therefore the mainstay of therapy for this type of amyloidosis involves chemotherapy aimed at eradicating these cells.
Oral or intravenous melphalan and dexamethasone, often combined with autologous stem cell support, have been used for many years.
Both drugs are equally effective, but the treatment and side effects are different. High dose melphalan with stem cell support is a course of treatment that often includes a 2-3 week hospital stay and several months of recovery. Use of oral melphalan in monthly courses is less toxic but is associated with a higher risk of developing leukemia.
Newer drugs active against multiple myeloma (another abnormal plasma cell disease), such as bortezomib or lenalidomide, are also very effective against AL and have been shown to provide some benefit to patients with recurrent disease. Often these drugs are included in pre-treatment.
Currently, most patients not using high-dose melphalan with stem cell support receive advanced therapy. The combination of bortezomib, cyclophosphamide and dexamethasone is associated with good tolerability and rapid responses. Treatment of amyloidosis for any patient should be tailored individually, taking into account the specifics of the situation.
The two most important factors for long-term survival with AL are the presence/extent of cardiac involvement and hematologic response to therapy.
There are several new drugs designed to stimulate the resorption of amyloid from affected organs. Their use may provide the ability to treat diseased organs directly. The most advanced of these studies is with NEOD001, which showed some benefit in patients whose underlying plasma cell disease had already been treated. The method is currently being studied in combination with early-stage bortezomib-based therapy.
Supportive care (treatment of congestive heart failure, attention to nutrition, treatment of autonomic neuropathy, etc.) is a very important element of drug management. Given the complexity of the disease, it is recommended that treatment be carried out in a specialized amyloidosis center or, at the very least, the patient should undergo an initial assessment in such a medical facility with continued treatment in the community.
Familial amyloidosis is treated, if possible, by removing the underlying cause of abnormal TTP production. Because the liver is the dominant source, organ transplantation is now the preferred choice in carefully selected patients whose disease is at acceptable stages of development. Tafamidis is a drug recently approved for the treatment of familial amyloid polyneuropathy. This drug is being tested in ongoing trials for other forms of the disease. Patisiran and revusiran are also being tested for their effects on the ATTR form of amyloidosis, with a focus on reducing levels of TTR, which forms amyloid.
Consultation with a geneticist is recommended for all individuals with hereditary amyloidosis and their family members.
In senile amyloidosis, therapy is favorable, but for both this form of the disease and ATTR, pharmacological treatments aimed at stabilizing the transthyretin molecule to prevent amyloid formation are being actively studied.
Secondary treatment of amyloidosis is based on the treatment of the main disease. For example, kidney transplantation may be performed for renal failure due to secondary amyloidosis.
In March 2015, the US Drug Administration (FDA) approved a medical device called Aheresis Column Lixelle Beta 2-microglobulin for the treatment of dialysis-associated beta2-microglobulin amyloidosis. The device works by removing beta-2-microglobulin from the blood.
Tests and diagnostics
Considering that amyloidosis can be disguised under different masks due to the excessive diversity of clinical symptoms, diagnosing the disease is considered very difficult. To assess the functional state of internal organs, examination methods such as:
- ECG;
- EchoCG;
- Ultrasound;
- Radiography;
- Sigmoidoscopy;
- FGDS.
Amyloidosis, which was suspected based on clinical symptoms and laboratory parameters, requires confirmation by analyzing the pathological substrate - amyloid. It is for this purpose that paint samples are used. According to one of the existing modifications, the dye (Congo red, Evans blue) is administered intravenously and accumulates in amyloid masses, thereby reducing its concentration in the blood.
Another diagnostic method is the subcutaneous injection of a 1% freshly prepared solution of methylene blue in a volume of 1 cubic centimeter into the subscapular area. Next, the color of the urine is assessed. The color of urine does not change if the amyloid masses have captured and accumulated the dye. In this case, the analysis is considered positive and the diagnosis confirmed. However, if the color of the urine remains unchanged and the test is negative, the diagnosis cannot be excluded.
The main diagnostic method is a biopsy of the affected organ (liver, kidney, etc.). In 90-100% of cases a positive result is achieved. The stronger the infiltration of the target organ by amyloid, the easier it is to identify the protein and establish an accurate diagnosis. Initially, biopsies are taken from the rectum or from the mucous membranes of the oral cavity in the area of 3-4 molars.
If AL amyloidosis is suspected, biopsy . It is also possible to perform an aspiration biopsy of subcutaneous fat from the anterior abdominal wall (sensitivity level reaches 50%). A biopsy of periarticular tissue is performed if dialysis amyloidosis is suspected.
Iodine-labeled serum P-component scintigraphy is now very common This method allows you to clearly track the dynamics of tissue deposits during the treatment process. Diagnostically, it is important not only to identify amyloid in tissues, but also to type it using colorful methods and antisera (mono- and polyclonal antibodies) to the main proteins of amyloid fibrils.
All methods for diagnosing amyloidosis today
Diagnosis of the disease in question involves the following activities:
- Conversation with the patient about the duration of anxiety symptoms . At this stage, it is important to find out whether any infectious pathology occurred shortly before the onset of complaints.
- Analysis of the patient's medical history and living/working conditions . The doctor is interested in the presence of rheumatic diseases, syphilis, tuberculosis, osteomyelitis, etc. in the past.
- Initial examination of the patient : measurement of blood pressure and pulse; study of the condition of the skin and mucous membranes, tongue; palpation of the abdomen.
- Clinical testing of blood and urine samples. With amyloidosis, the level of hemoglobin and leukocytes increases, and the ESR accelerates. The concentration of platelets in the blood decreases. Urine with amyloidosis of the kidneys and gastrointestinal tract contains protein, leukocytes, blood, and mucus.
- Biochemical blood test for the level of proteins, creatinine, urea, and cholesterol.
- Ultrasound diagnostics of internal organs. Makes it possible to evaluate their structure and quality of functioning. Using this technique, you can study the structure of the liver, spleen, gastrointestinal tract, heart, and large vessels.
- Barium X-ray. Relevant for suspected intestinal amyloidosis. Using this technique, it is possible to assess the level of intestinal patency, identify narrowed areas, and defects in the intestinal structure.
- A biopsy of the affected organ is the gold standard in diagnosing amyloidosis. The required material can be collected from the mucous membrane of the stomach, lymph node, kidney, rectum, gums, liver, muscle tissue, spleen, and other organs.
Rate
—
Treatment and medications
The main therapy is aimed at suppressing the synthesis and delivery of precursor proteins from which amyloid is formed.
Treatment of AA amyloidosis
Therapy is aimed at treating the disease that led to amyloidosis. Chemotherapy, antibiotics, surgery, etc. are used.
Preference is given to 4-aminoquinoline derivatives ( Rezokhin , Plaquenil , Delagil ). The action of medications is aimed at suppressing the synthesis of amyloid fibrils at the earliest stages of amyloidogenesis by inhibiting several enzymes. The drug Delagil in a dose of 0.25 g is prescribed for a long time (several years).
The protein fibrils that form amyloid contain a huge number of SH-free sulfhydryl groups, which take an active part in the aggregation of proteins into stable structures. To block them, 5% Unithiol intramuscularly in a dose of 3-5 ml daily with a gradual increase in dosage to 10 ml per day for 30-40 days. A repeated course is carried out 2-3 times a year.
It is mandatory to consume 100-150 g of cooked or raw liver daily for 6-12 months. The use of liver hydrolysates, such as Sirepar . 40 g of liver corresponds to 2 ml of Sirepar . The recommended regimen involves alternating intramuscular administration of Sirepar twice a week, 5 ml (2-3 months) with the consumption of raw liver for 1-2 months.
Immunomodulators have proven themselves well:
- Levamisole or Decaris ;
- T-activin;
- Timalin.
Dimexide has a positive effect due to its absorbable effect. The medication is used in the form of 10-20% orally, 10 mg, course for 6 months.
Colchicine has an antimitotic effect and is used for periodic illness. The positive effect of the drug is explained by its ability to slow down amyloidogenesis. With timely administration, the development of renal amyloidosis, which is considered the most dangerous complication, can be prevented. Colchicine is prescribed for a long time, and in some cases for life.
Treatment of AL amyloidosis
The goal of treatment is to reduce the production of precursors - light chains of immunoglobulins, which is why polychemotherapy is indicated. The standard regimen includes Prednisolone and Melphalan . More aggressive regimens contain Doxorubicin , Vincristine , Cyclophosphamide .
To improve the functioning of T-suppressors, Levamisole and other immunomodulators are prescribed. For ATTR amyloidosis, liver transplantation is indicated.
For dialysis amyloidosis and the AB2M form, high-flow hemodialysis with immunosorption and hemofiltration is prescribed. Thanks to timely procedures, it is possible to reduce the level of beta2-microglobulin. In critical cases, kidney transplantation is indicated.
Prescribed therapy does not always produce the expected effect due to late diagnosis and the involvement of several organs in the pathological process. Timely, as early as possible diagnosis, based on knowledge of all the symptoms of amyloidosis, is of decisive importance.
Medicines
- Rezokhin.
- Plaquenil.
- Delagil.
- Sirepar.
- Unithiol.
Procedures and operations
Transplantation of a donor organ is the only possible way to save the life of a patient with organ failure that has developed against the background of amyloidosis. It is important to understand that transplantation is only a symptomatic treatment method that is not able to eliminate the true cause and slow down the progression of amyloidosis. Even after transplantation, recurrence of the disease is possible.
What can be transplanted for amyloidosis:
- heart;
- kidney;
- skin;
- liver tissue.
A donor organ can be obtained either from a living donor or from a person who has been diagnosed with brain death, but at the same time retains the functional activity of the internal organs. To date, an artificial heart has been developed, which is presented in the form of a fully mechanized device that pumps blood in the human body. After transplantation, lifelong use of immunosuppressants is prescribed, which helps prevent rejection of the “foreign” tissue or organ.
In adults
As mentioned above, the disease can occur in old age. It also occurs among middle-aged people. How to cure the disease at the secondary stage? The diet of an adult is important. This diet applies to adults.
- more carbohydrates;
- salt restriction;
- a significant amount of vitamins
Drug therapy is used. Hemodialysis is prescribed. Diuretics to reduce swelling. Medicines that lower blood pressure. Plasma transfusion or kidney transplant. It is worth considering that treatment of edema and plasma transfusion are not sufficient; this gives only a temporary effect. If renal amyloidosis is caused by an infectious disease, then it is important to eliminate its spread and further relapses.
go to top
During pregnancy
Pregnancy with amyloidosis is allowed only if the functional activity of the woman’s vital organs allows her to bear and give birth to a healthy child. Otherwise, pregnancy can cause death not only for the fetus, but also for the mother herself.
Certain local forms of amyloidosis do not prevent the onset and gestation of pregnancy. Childbirth can proceed without complications and result in the birth of a healthy baby when amyloid protein accumulates in only one specific organ or tissue (intestinal wall, muscle tissue). In the generalized form, the prognosis for the mother and fetus is determined by the reserves of the affected organ and the duration of the disease.