Мукополисахаридоз – наследственное заболевание соединительной ткани, связанное с нарушением обмена гликозаминогликанов, для которого характерен специфический внешний вид лица, аномалии глаз, костей, селезенки, печени. В некоторых случаях заболевание может сопровождаться задержкой умственного развития.
Существуют различные типы мукополисахаридоза, развитие каждого из которых обусловлено определенной генетической аномалией, которая оказывает влияние на синтез специфического фермента.
Существующие методы лечения мукополисахаридоза имеют низкий уровень эффективности.
Причины мукополисахаридоза
Причиной развития мукополисахаридоза является нарушение ферментативного катализа гликозаминогликанов в лизосомах. При мукополисахаридозе нарушается процесс расщепления и сохранения мукополисахаридов, которые являются основными компонентами соединительной ткани. Избыток мукополисахаридов проникает в кровь и накапливается в тканях. Поэтому данное заболевание относят к болезням накопления.
В настоящее время выявлено 10 генетических типов мукополисахаридоза, четыре из которых возникают при нарушении активности гликозидаз, пять – сульфатаз, один развивается в случае дефицита трансферазы.
Заболевание наследуется по аутосомно-рецессивному и рецессивному, сцепленному с Х-хромосомой типам.
Что такое мукополисахаридоз?
Мукополисахаридозы (сокр. МПС)— это группа наследственных метаболических заболеваний, вызванных отсутствием или неисправностью определенных ферментов, необходимых организму для расщепления молекул, называемых гликозаминогликанами, — длинных цепочек сахаров (углеводов) в каждой из наших клеток. Эти клетки помогают строить кости, хрящи, сухожилия, роговицу, кожу и соединительную ткань. Гликозаминогликаны (ранее называемые мукополисахаридами) также содержатся в жидкости, которая смазывает суставы.
Больные мукополисахаридозом либо не вырабатывают достаточно одного из 11 ферментов, необходимых для расщепления этих сахарных цепей на белки и более простые молекулы, либо производят ферменты, которые не работают должным образом. Со временем эти гликозаминогликаны накапливаются в клетках крови, головном и спинном мозге, а также в соединительных тканях. Результатом является необратимое прогрессирующее повреждение клеток, которое влияет на внешний вид человека, физические способности, функционирование органов и систем и, в большинстве случаев, на умственное развитие. Симптомы могут быть похожими или различаться для разных типов расстройства.
Мукополисахаридозы классифицируются в рамках более широкой группы заболеваний, называемых лизосомными болезнями накопления. Это заболевания, при которых большое количество молекул, которые обычно распадаются на более мелкие части во внутриклеточных компартментах, называемых лизосомами, накапливаются в вредных количествах в клетках и тканях организма, особенно в лизосомах. Основная функция лизосом — переваривать нефункциональные клетки и другие материалы (включая бактерии и клеточный мусор).
Другой лизосомной болезнью накопления, которую часто путают с мукополисахаридозом, является муколипидоз. При этом заболевании в дополнение к более мелким углеводам, называемым сахарами, накапливается чрезмерное количество жировых веществ, известных как липиды (еще один основной компонент живых клеток). Больные муколипидозом могут иметь общие клинические особенности, связанные с мукополисахаридозами (определенные черты лица, аномалии костной структуры и повреждение головного мозга), однако эти болезни разные.
Типы и симптомы мукополисахаридоза
Выделяют следующие типы мукополисахаридоза:
- I тип или синдром Гурлера;
- II тип или синдром Гунтера;
- III тип или синдром Санфилиппо;
- IV тип или синдром Моркио;
- V тип или синдром Шейе;
- VI тип или синдром Марото-Лами;
- VII тип или синдром Слая;
- VIII тип или синдром Ди Ферранте.
Симптомы мукополисахаридоза обнаруживаются у ребенка на первом году жизни и уже к 12-24 месяцам они становятся довольно выраженными.
У таких детей наблюдаются:
- Череп в форме лодочного киля;
- Шумное дыхание ртом;
- Грубые черты лица;
- Отставание в росте;
- Деформации скелета;
- Постепенное развитие сгибательных контрактур;
- Увеличение размеров живота;
- Пупочные и паховые грыжи;
- Гидроцеле;
- Изменения со стороны глаз: помутнение и увеличение размеров роговицы, врожденная глаукома, атрофия сосков зрительных нервов, застойные явления на глазном дне, пигментная дистрофия сетчатки;
- Снижение слуха;
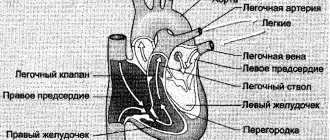
- Изменения со стороны сердца;
- Умственная отсталость;
- Повышенный тонус мышц, нарушение координации движений, параличи.
При I типе заболевания симптомы мукополисахаридоза при рождении отсутствуют. Первые симптомы мукополисахаридоза в виде ограниченного разгибания пальцев на руках наблюдаются только в 3-6 лет. Со временем уменьшается объем движений в других суставах рук. К периоду полового созревания все симптомы проявляются особенно ярко.
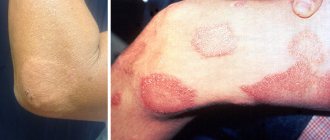
II тип мукополисахаридоза чаще встречается у мальчиков. Для больных характерны: скафоцефалия, грубые черты лица, низкий грубый голос, шумное дыхание, частые ОРВИ. В 3-4 года возникают нарушения координации движений. Наблюдается прогрессирующая тугоухость, остеоартриты, узелковые поражения кожи спины.
При наличии синдрома Санфилиппо (III тип) ребенок в течение 3-5 лет нормально развивается, хотя иногда могут наблюдаться затрудненное глотание и неуклюжая походка. После трех лет у ребенка начинает развиваться апатия, возникает задержка психомоторного развития, нарушается речь, грубеют черты лица, возникает недержание кала и мочи, ребенок перестает узнавать окружающих. Такие дети умирают в возрасте 10-20 лет из-за интеркуррентных инфекций.
IV тип мукополисахаридоза характеризуется появлением первых симптомов в 1-3 года. Наблюдаются: резкая задержка роста, непропорциональное телосложение, грубые черты лица, кифоз или сколиоз, деформация грудной клетки; контрактуры в плечевых, локтевых, коленных суставах; плоскостопие; снижение мышечной силы; снижение слуха; дистрофия роговицы. Больные не доживают до 20-летнего возраста по причине сердечно-легочной недостаточности.
Для синдрома Шейе (V тип) характерен низкий рост, уплощенная переносица, короткая шея, контрактуры суставов, гипотония мышц конечностей, вегетативная лабильность, снижение сухожильных рефлексов, значительное помутнение роговиц.
Синдром Марото-Лами (VI тип) впервые проявляется после 2 лет. Он характеризуется отставанием в росте, грубыми чертами лица, бочкообразной грудной клеткой, малыми размерами верхней челюсти, короткой шеей, сгибательными контрактурами суставов рук. Интеллект больных не страдает.
Мукополисахаридоз VII типа устанавливают лишь при детальном биохимическом исследовании.
Синдром Ди Ферранте (VIII тип) схож с синдромом Моркио, но отличается выраженной задержкой интеллектуального и психомоторного развития.
Орфанные заболевания в практике педиатра: мукополисахаридоз
С 1 января 2012 г. в силу вступил новый Федеральный закон от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации», в котором впервые на государственном уровне введено понятие редких (орфанных) заболеваний. В список орфанных болезней на сегодняшний день внесено 230 наименований, однако в случае выявления новых болезней он будет пополняться. По данным Формулярного комитета Российской академии медицинских наук (РАМН), насчитывается около 300 тыс. россиян с этими болезнями.
Редкие (орфанные) заболевания — это встречающиеся с определенной частотой жизнеугрожающие или хронические прогрессирующие заболевания, при отсутствии лечения приводящие к смерти или пожизненной инвалидизации пациентов.
В разных странах определение и перечень орфанных болезней принимаются на государственном уровне, единого определения для них не существует, также как нет единого критерия отнесения заболеваний к этой группе.
В нашей стране к орфанным относятся заболевания, которые имеют распространенность не более 10 случаев заболевания на 100 000 населения.
Сегодня в мире насчитывается около 7000 редких заболеваний. Примерно половина из них обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте.
Одним из заболеваний, включенным в группу орфанных, является мукополисахаридоз (МПС) (код по МКБ-10 Е76.1 — «нарушение обмена гликозаминогликанов», Постановление Правительства № 403 от 26 апреля 2012 г.).
Мукополисахаридоз — это группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов (ГАГ) (кислых мукополисахаридов) в результате генетически обусловленной неполноценности 1 из 11 известных ферментов, участвующих в их расщеплении. Относится к лизосомным болезням накопления (табл.).
Для МПС характерно полисистемное поражение: прогрессирующие психоневрологические нарушения, гепатоспленомегалия, сердечно-легочные расстройства, костные деформации [1–3].
В Московской области зарегистрированы и получают лечение 3 ребенка с МПС: 1 девочка с МПС I типа (синдром Гурлер–Шейе) и 2 мальчика с МПС II типа (синдром Хантера). Еще два ребенка в настоящее время находятся в стадии обследования, уточняется тип МПС.
Представляем клиническую и лабораторную характеристику заболевания на примере МПС II типа.
Мукополисахаридоз II типа (синонимы: недостаточность фермента лизосомной идуронат-2-сульфатазы (a-L-идуроносульфатсульфатазы), синдром Гунтера (Хантера) — сцепленное с Х-хромосомой рецессивное заболевание, возникающее в результате снижения активности лизосомной идуронат-2-сульфатазы, участвующей в метаболизме ГАГ. Возникает почти исключительно у мальчиков (XY). У гетерозиготных женщин клинических проявлений синдрома Хантера, как правило, не наблюдается («носители»). К настоящему моменту описано лишь 2 случая заболевания у девочек, связанных с инактивацией второй, нормальной, Х-хромосомы.
Это панэтническое заболевание, частота встречаемости в мире — около 1 на 75 000 живых новорожденных мальчиков. Частота заболевания в популяции варьирует от 1 на 165 000 (Австралия) до 1 на 34 000 (Израиль). Развитие МПС II типа обусловлено мутациями в структурном гене лизосомной идуронат-2-сульфатазы — IDS, расположенном на длинном плече Х-хромосомы в локусе Xq28. В настоящее время описано более 300 различных мутаций в гене IDS. Более 50% мутаций составляют точечные мутации, около 26% — мелкие делеции и вставки, 11% — крупные делеции и перестройки гена IDS. Для пациентов России ДНК-анализ гена IDS показал, что крупные делеции и перестройки гена IDS составляют только 5,4% числа найденных мутаций.
Клинический фенотип гетерогенен и довольно условно подразделяется на тяжелую и легкую формы, различающиеся по тяжести клинических фенотипов. У пациентов с тяжелой формой наблюдают сходные с МПС I типа (синдромом Гурлер) клинические симптомы, но заболевание прогрессирует медленнее (рис.).
Заболевание манифестирует в возрасте от 1 до 3 лет. Отмечается изменение черт лица по типу гаргоилизма, задержка роста, признаки множественного костного дизостоза, огрубление и утолщение кожи, прогрессирующее снижение интеллекта. Специфичными для данного типа МПС являются изменения на коже спины, груди, шеи цвета слоновой кости, «монголоидные пятна» в пояснично-крестцовой области. Нарушения функции органов пищеварения проявляются в виде гепатомегалии, хронической диареи. Среди неврологических нарушений преобладают симптомы прогрессирующей сообщающейся гидроцефалии, спастическая параплегия в результате компрессии спинного мозга и прогрессирующая тугоухость. У детей с синдромом Хантера отмечается тугоподвижность крупных и мелких суставов. Прогрессируют сердечно-легочные расстройства. Летальный исход обычно наступает на втором десятилетии жизни.
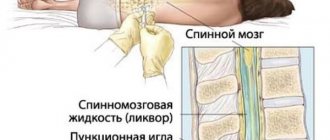
Для подтверждения МПС II типа проводится определение уровня экскреции ГАГ с мочой и измерение активности лизосомной идуронат-2-сульфатазы в лейкоцитах или культуре кожных фибробластов. В случае заболевания в моче возрастает суммарная экскреция ГАГ.
Проведение ДНК-анализа — длительная и сложная диагностическая процедура, позволяющая определить молекулярные дефекты, приводящие к болезни Хантера, наиболее часто используется для определения носительства и пренатальной диагностики в семьях, имеющих больного ребенка. Применяются методы косвенной ДНК-диагностики, основанной на исследовании локусов Х-хромосомы, расположенных близко к гену IDS.
Кроме того, пренатальная диагностика проводится путем измерения активности идуронат-2-сульфатазы в биоптате ворсин хориона на 9–11 неделе беременности или определения спектра ГАГ в амниотической жидкости на 20–22 неделе беременности.
Наиболее часто дифференциальная диагностика проводится внутри группы МПС, а также с другими лизосомными болезнями накопления (муколипидозами, галактосиалидозом, ганглиозидозом и др.).
Основным подходом к лечению больных с МПС является проведение пожизненной ферментозаместительной терапии. При МПС II типа используется препарат идурсульфаза (Элапраза) производства , США, зарегистрированный в странах Европы, США, России для лечения мукополисахаридоза II типа (болезнь Хантера). Препарат вводится еженедельно, внутривенно, капельно, медленно в дозе 2 мг/кг.
В качестве клинического примера приводим выписку из истории болезни.
Никита Б., дата рождения 11.12.2011.
Анамнез жизни. Ребенок от второй беременности (первая беременность окончилась выкидышем на раннем сроке), протекавшей на фоне маловодия, гипоксии плода, угрозы прерывания, отеков. Роды срочные, самостоятельные. Вес при рождении 4040 г, рост 58 см. С рождения на грудном вскармливании.
Формула развития: голову держит с 2 мес, переворачивается с 5 мес, сидит с 9 мес, ходит с 1 г 1 мес. К году — 5 слов. Перенесенные заболевания — острая респираторная вирусная инфекция 2 раза, трахеит.
Анамнез заболевания. С рождения — водянка оболочек яичек, с 5 мес — пахово-мошоночная грыжа, в 7 мес диагностирован кифоз поясничного отдела позвоночника, в 12 мес по данным электрокардиографии (ЭКГ) — синдром ранней реполяризации желудочков, неполная блокада правой ножки пучка Гиса (НБПНГ), в 1 г 3 мес проведена эхокардиография (Эхо-КГ), выявлена аневризма межпредсердной перегородки (МПП).
В 1 г 4 мес ребенок обследован в МГНЦ РАМН, заподозрен и подтвержден биохимическим и молекулярно-генетическим методами МПС II типа.
При поступлении в клинику состояние ребенка средней тяжести. Вес 15 кг. Рост 93 см. Волосы жесткие, тусклые, макростомия, макроглоссия, короткая шея, легкие черты гаргоилизма, камподактилия 4,5 пальца справа, широкое пупочное кольцо, кифоз поясничного отдела позвоночника, килевидная деформация грудной клетки, пахово-мошоночная грыжа слева, кожа — плотная на ощупь. Гипертрофия миндалин 2–3 ст. Дыхание через нос не затруднено. Дыхание везикулярное. Тоны сердца ритмичные, акцент второго тона. Частота сердечных сокращений (ЧСС) 120 уд./мин. Живот увеличен в объеме, печень +3,5 см, +4 см верхняя треть. Селезенка не пальпируется. Стул оформленный. Дизурических явлений нет.
Неврологический статус: в сознании, гиперактивен, быстро возбудим, говорит отдельные слова. Общемозговых, менингеальных знаков нет. Окружность головы = 50 см. Глазные щели OD = OS. Зрачки OD = OS. Движение глазных яблок в полном объеме. Низкий тембр голоса. Быстро истощается, ходит самостоятельно, легкая атаксия. Мышечная гипотония. Сухожильные рефлексы живые.
Общий анализ крови и мочи без патологических изменений.
Биохимический анализ крови — умеренное повышение уровня аланинаминотрансферазы (АЛТ) (42 Ед/л), аспартатаминотрансферазы (АСТ) (49 Ед/л).
ЭКГ: вертикальное положение электрической оси сердца (ЭОС), выраженная синусовая тахикардия 182–200 на фоне беспокойства. В ортостазе ЧСС 111–166 уд./мин.
Ультразвуковое исследование (УЗИ) брюшной полости: печень увеличена, правая доля 100 мм, левая — 45 мм. Структура однородная, стенки сосудов и протоков уплотнены. Эхогенность повышена. Поджелудочная железа 14 × 8 × 15 мм, структура неоднородная, эхогенность не изменена. Желчный пузырь — форма обычная, просвет чистый, стенки не утолщены. Селезенка не увеличена, 62 × 32 мм. Структура однородная. Почки без патологических изменений. Яички в мошонке, вагинальный отросток брюшины расширен с обеих сторон до 3 мм.
Электроэнцефалография (ЭЭГ): диффузные общемозговые изменения биоэлектрической активности по органическому типу в виде доминирования дельта-активности частотой 3–4 Гц по всем отделам конвекса безградиентно в сочетании с отдельными группами низкочастотных тета-колебаний. Эпилептиформная активность, фотопароксизмальная реакция не выявлены.
На основании комплексного исследования установлен диагноз: «Мукополисахаридоз II типа (синдром Хантера). Дегенеративное заболевание нервной системы. Задержка речевого развития. Синдром гипервозбудимости. Множественный костный дизостоз. Порок развития L1-L2 позвонков, врожденный кифоз. 2-стороннее гидроцеле. Пупочная грыжа».
По жизненным показания ребенку назначено внутривенное введение Элапразы в дозе 0,5 мг/кг еженедельно (15 кг × 0,5 = 7,5 мг на введение).
Ребенок с июля 2013 г. (возраст 1 г 7 мес) получает ферментозаместительную терапию в условиях детского отделения ГБУЗ МО МОНИКИ. В комплексную терапию включены занятия с логопедом, дефектологом, курсы физиотерапии, лечебная физкультура, L-карнитин (Элькар), Кудесан, глицин, церебролизин, Магне В6.
Литература
- Meikle P. J. et al. Prevalence of lysosomal storage disorders // JAMA. 1999. 281: 249–254.
- Neufeld E. F., Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al (eds). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001: 3421–3452.
- Fenton C. L., Rogers W. Mucopolysaccharidosis type II. eMedicine Journal . 2006. www.emedicine.com/ped/topic1029.htm. Accessed on April 3, 2006.
Т. А. Бокова1, кандидат медицинских наук Е. В. Лукина Н. В. Шестериков
ГБОУ МО МОНИКИ им. М. Ф. Владимирского, Москва
1 Контактная информация
Диагностика мукополисахаридоза
Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления экскреции гликозаминогликанов с мочой, исследования активности ферментов в фибробластах кожи.
Диагностировать мукополисахаридоз можно еще до рождения ребенка, используя для анализа амниотическую жидкость или ворсины хориона.
Затронутые группы населения
Распространенность всех форм мукополисахаридоза оценивается в 1 случай на 25 000 рождений. Однако в связи с тем, что мукополисахаридозы, особенно легкие формы заболеваний, часто остаются нераспознанными, эти заболевания недодиагностированы или диагностируются неправильно, что затрудняет определение их истинной частоты среди населения в целом.
Оценки для конкретных типов мукополисахаридоза колеблются от: 1 на 100 000 для синдрома Гурлера; 1 на 500 000 для синдрома Шейе; 1 из 115 000 страдает синдромом Гурлера-Шейе; 1 из 70 000 синдромом Санфилиппо; 1 из 200 000 синдромом Моркио; и менее 1 из 250 000 человек синдромом Слая. Синдром Хантера встречается преимущественно у мужчин. В крайне редких случаях сообщалось о болезни женщин. Заболеваемость синдромом Хантера оценивается в 1 из 100 000–150 000 новорожденных мальчиков.
Выявлено более 40 различных лизосомных болезней накопления.
Мукополисахаридоз 1 типа. Диагностика
Лабораторные исследования
Следующие исследования показаны пациентам с подозрением на мукополисахаридоз 1 типа:
- Лимфоциты в мазках крови могут быть рассмотрены на наличие аномальных цитоплазматических структур.
- Мочевые уровни мукополисахаридов, дерматансульфата и гепарансульфата увеличиваются.
- Уровни L-идуронидазы могут быть определены в культуре фибробластов и в лейкоцитах.
- Пренатальная диагностика уровня фермента может быть также выполнена при анализе амниотических клеток и ворсинок хориона.
Визуализация
- В тяжелых случаях мукополисахаридоза 1 типа, рентгенография скелета (особенно позвоночника) может быть полезной для обнаружения деформаций костей позвоночника.
- Эхокардиография полезна в определении характера сердечных осложнений.
Примечания
- OMIM 607014 (англ.)
- OMIM 607016 (англ.)
- OMIM 607015 (англ.)
- OMIM 309900 (англ.)
- OMIM 252900 (англ.)
- OMIM 252920 (англ.)
- OMIM 252930 (англ.)
- OMIM 252940 (англ.)
- OMIM 253000 (англ.)
- OMIM 253010 (англ.)
- OMIM 253200 (англ.)
- OMIM 253220 (англ.)
- ↑ 123
Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил.
Глава 316. Лизосомные болезни накопления (с. 250—273)
. med-books.info. Проверено 30 ноября 2014. - ↑ 12
Внутренние болезни: учебник: в 2 т. / под ред. В. С. Моисеева, А. И. Мартынова, Н. А. Мухина. — 3-е изд., испр. и доп. — 2013. — Т.2. — 896 с.: ил..
Часть XIII. Наследственные болезни накопления
. vmede.org. Проверено 26 января 2015.
Стандартные методы лечения
В настоящее время лекарства от этих расстройств не существует. Медицинская помощь направлена на лечение системных заболеваний и улучшение качества жизни человека. Изменения в питании не предотвратят прогрессирование болезни.
Хирургическое вмешательство может помочь вывести излишки спинномозговой жидкости из головного мозга и освободить нервы и нервные корешки, сдавленные скелетными и другими аномалиями. Трансплантация роговицы может улучшить зрение у людей со значительным помутнением роговицы. Удаление миндалин и аденоидов может улучшить дыхание у людей с обструктивными нарушениями дыхательных путей и апноэ во сне. Некоторым людям может потребоваться хирургическое введение эндотрахеальной трубки для облегчения дыхания.
Ферментно-заместительная терапия в настоящее время используется для лечения МПС I, МПС II, МПС IVA, МПС VI и МПС VII типов, а также тестируется при других расстройствах, связанных с мукополисахаридозами. Заместительная ферментная терапия включает внутривенное введение раствора, содержащего фермент, который отсутствует в организме. Он не лечит неврологические проявления заболевания, но доказал свою эффективность в уменьшении неневрологических симптомов и боли.
Трансплантация костного мозга (ТКМ) и трансплантация пуповинной крови (ТПК) имели ограниченный успех в лечении мукополисахаридозов. Патологические физические признаки, за исключением тех, которые влияют на скелет и глаза, могут быть улучшены, но неврологические исходы могут быть разными. ТКМ и ТПК являются процедурами с высоким риском и обычно выполняются только после тщательного обследования и консультации членов семьи.
Физическая терапия и ежедневные упражнения могут отсрочить проблемы с суставами и улучшить подвижность.
Лечение мукополисахаридоза
Важное значение в лечении ребенка с мукополисахаридозом имеет раннее установление диагноза с определением клинико-лабораторного типа данной патологии, так как каждый вариант течения заболевания требует индивидуального подхода к применению целесообразного метода лечения. В определении тактики ведения и лечения пациента, страдающего мукополисахаридозом того или иного типа принимает участие группа специалистов, включающая педиатра-неонатолога, специалиста пластической хирургии, ортопеда, нейрохирурга, офтальмолога и отоларинголога.
Для лечения используются гормональные препараты, такие как глюкокортикоиды, кортикотропин и тиреоидин. Данные средства способствуют лишь временному улучшению состояния больного. Витамин А, введение декстрана-70, а также переливание препаратов крови плазмы также используются для лечения мукополисахаридоза, однако как и гормональные препараты способствует лишь временному улучшению состояния.
В мировой врачебно практике при мукополисахаридозе есть несколько методик применяемого лечения:
- симптоматическое:
- медикаментозное;
- хирургическoe;
- физиотерапевтическое — электрофорез лидазы на область пораженных суставов, магнитотерапия, парафиновые аппликации, лазерная пунктура, ЛФК с воздействием на позвоночник и суставы, общий массаж, санация хронических очагов инфекции носоглотки и полости рта.
- заместительная терапия;
- трансплантация стволовых клеток;
- антиглаукоматозные операции;
- грыжесечение;
- аденотонзиллэктомия;
- трахеостомия;
- шунтирование при гидроцефалии;
- операции карпального туннельного синдрома;
- протезирование клапанов сердца.
Лечение состоит в назначении таких гормональных препаратов, как:
- АКТГ — с целью для уменьшить синтез мукополисахаридов;
- тиреоидин;
- глюкокортикоиды;
- кортикотропин;
- преднизолон.
Тем не менее, употребление гормональных препаратов дают всего лишь временные результаты.
Типы
Существуют основные симптомы данного заболевания, к которым относятся:
- выраженные деформации скелета;
- деформации костей черепа, ушных раковин, искривление зубов;
- огрубение черт лица;
- задержка роста;
- часто наблюдается задержка умственного развития;
- снижение или полное ограничение подвижности суставов;
- частое развитие грыж (разной локализации);
- снижение слуха и зрения;
- поражение внутренних органов и предрасположенность к частым инфекционным заболеваниям.
В зависимости от того, насколько выраженными являются физические и психологические нарушения, выделяют семь типов такого заболевания, как мукополисахаридоз. При этом основными, которые встречаются чаще других, являются четыре типа.
Мукополисахаридоз 1 типа
Мукополисахаридоз 1 типа (болезнь Гурлера). Данная патология является самой быстропрогрессирующей, протекающей с выраженной симптоматикой. Характерным симптомом для неё является деформация черепа с развитием грубых черт лица. К двум годам клиническая картина уже полностью выражена и представлена серьёзными деформациями скелета, нарушением работы сердца, печени и селезёнки, а также выявлением множественных грыж. Прогноз заболевания крайне неблагоприятен.
Мукополисахаридоз 2 типа
Что это такое? Эта разновидность мукополисахаридоза еще называется болезнью Хантера. Патологией чаще всего страдают мальчики в возрасте 2-3 лет. По клиническим проявлениям заболевание похоже на мукополисахаридоз типа IH – при этом наблюдаются:
- скароцефалия;
- грубые черты лица;
- низкий тембр голоса;
- затруднение дыхания – появляется из-за деформации костей лицевого скелета.
При дальнейшем прогрессировании заболевания присоединяются следующие признаки:
- снижается интеллект;
- нарушается координация движений;
- настроение у такого пациента неустойчивое – отмечается так называемая эмоциональная лабильность – от приступов смеха до плача. Такие резкие перепады настроения могут появиться в любое время.
При физикальном обследовании определяется следующее:
- выявляется незначительное увеличение селезенки и печени;
- на коже спины определяются специфические узелки.
При инструментальном обследовании выявляются:
- помутнение роговицы, которое прогрессирует;
- нарастающая тугоухость.
Из всех инструментальных методов диагностики наиболее информативной является рентгенография различных фрагментов опорно-двигательного аппарата – черепа, рудной клетки, таза и так далее. При этом рентгенологические признаки те же, что и при мукополисахаридозе типа I-S.
Пациенты с таким диагнозом склонны к развитию заболеваний дыхательной системы:
- трахеитов;
- бронхитов;
- пневмоний.
Следует учесть, что мукополисахаридоз типа II может развиваться в виде двух вариантов:
- неблагоприятного (вариант А);
- благоприятного (вариант В).
3 тип
Мукополисахаридоз III типа (синдром Санфилиппо) характеризуется тяжелыми неврологическими симптомами. К ним относятся прогрессирующая деменция, агрессивное поведение, гиперактивность, судороги, некоторая глухота и потеря зрения, а также невозможность спать более нескольких часов за раз. Мукополисахаридоз 3 типа воздействует на больных по-разному. Развитие умственных и моторных навыков может несколько замедленным. У больных наблюдается:
- заметное снижение возможности обучения в возрасте от 2 до 6 лет, а затем в конечном итоге утрачиваются языковые навыки и теряется часть слуха.
- Отдельные дети никогда не могут научиться говорить.
- Агрессивное поведение, гиперактивность, глубокая деменция и нерегулярный сон могут затруднить контроль за детьми, особенно теми, у кого сохраняется нормальная физическая сила.
- По мере прогрессирования болезни дети становятся все более неустойчивыми, и большинство из них не могут ходить в возрасте до 10 лет.
С возрастом становятся заметными складки кожи и изменения лицевых особенностей, костных и скелетных структур. Рост обычно останавливается к 10 годам. Другие проблемы могут включать сужение прохода дыхательных путей в горле и увеличение миндалин и аденоидов, что затрудняет глотание.
Ожидаемая продолжительность жизни в при III типе мукополисахаридоза чрезвычайно различна. Большинство больных живут до подросткового возрасте, а некоторые живут дольше, до двадцати или тридцати лет. Заболеваемость мукополисахаридозом 3 типа (для всех четырех подтипов в сочетании) составляет на около одного из 70 000 родов.
Мукополисахаридоз типа 4
Мукополисахаридоз типа IV (синдром Моркио, болезнь Моркио). Заболевание в 1929 г. независимо друг от друга впервые описали уругвайский педиатр Моркио (L. Morquio) и английский радиолог Брейлсфорд (J.F. Braiisford). Частота до 1: 40 000. Дети рождаются без признаков болезни.
Первые симптомы появляются в возрасте 1—3 года, и к 7—8 годам клиническая картина уже полностью выражена:
- Отмечаются резкая задержка роста (рост взрослого больного около 100 см),
- непропорциональное телосложение (относительно короткое туловище, микроцефалия, короткая шея),
- грубые черты лица,
- деформация грудной клетки (куриная, бочкообразная, килеобразная),
- кифоз или сколиоз грудного и поясничного отделов позвоночника.
- Питание снижено.
- Возникают контрактуры в локтевых, плечевых, коленных суставах, отмечается вальгусная деформация нижних конечностей, плоскостопие. Мышечная сила снижена.
В случае компрессии спинного мозга помимо мышечной гипотонии отмечается поражение пирамидной системы, возможно развитие параплегии, паралича дыхания. Кожа утолщена, ее тургор и эластичность снижены. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота. Нередко отмечается снижение слуха, дистрофические процессы в роговице. Почти у всех больных, доживших до 20 лет, развивается глухота. Интеллект не снижен.
Тип I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком становятся сгибательные контрактуры пальцев рук Дюпюитрена. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев.
У некоторых больных выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз 6 типа
Что это за болезнь? Мукополисахаридоз VI типа (иначе называемый синдромом Марото-Лами или болезнью Морото-Лами) – первая симптоматика данной формы начинает появляться у детей в возрасте после 2-х лет. Типичными особенностями являются отставание в росте, огрубление черт лица (такое же как и при синдроме Гулера, но не так сильно выраженное), маленькие размеры верхней челюсти, бочкообразная грудная клетка, чересчур короткая шея, укороченные ключицы.
Можно отметитьпоявление сгибательных контрактур суставов рук (больные не в состоянии самостоятельно поднимать руки вверх), с ходом времени возникают контрактуры и в суставах нижних конечностей, характерны нарушения походки. Довольно часто сюда же присоединяются частые острые респираторные инфекции.
Можно нередко выявить и грыжи, а также гепатоспленомегалию. В ряде случаев может развиться гидроцефалия, а также спастические параличи. Интеллектуальные способности больного при данной форме мукополисахаридоза не страдают. По степени выраженности симптоматики мукополисахаридоза заболевание Марото-Лами можно подразделить на типичную классическую форму (форму А) или более мягкую (форму В), при которой клинические проявления выражены не так сильно.
Другие типы мукополисахаридоза
Мукополисахаридоз 7 типа протекает, как тип 3, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа 8 по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Клиническая картина
В клинической практике все 12 известных мукополисахаридозов согласно фенотипу делят на две группы: Гурлер-подобный (10) и Моркио-подобный фенотип (2: синдром Моркио A и B)[14]:
| Фенотипы мукополисахаридоза[14]: | |
| Гурлер-подобный фенотип: | Моркио-подобный фенотип: |
| низкий рост с диспропорциональным строением скелета (относительно длинными конечностями, короткими туловищем и шеей); | диспропорциональная карликовость; |
| грубые черты лица (запавшее переносье, нередко экзофтальм, густые сросшиеся брови, полные губы, большой, нередко не помещающийся в полости рта язык); | грубоватые черты лица; |
| костные деформации (кифосколиоз, воронкообразная деформация грудной клетки); | килевидная деформация грудной клетки; |
| контрактуры крупных и мелких суставов; | гипермобильность межфаланговых и тугоподвижность крупных суставов; |
| мышечная гипотония; | «браслетки», «чётки», увеличение объёма коленных суставов и их вальгусная деформация; |
| наличие пупочной и пахово-мошоночной грыжи; | |
| гипертрофия лимфатического глоточного кольца; | |
| гипертрихоз; | |
| гепатоспленомегалия. | |
| Типична патология: | |
| центральной нервной системы (снижение интеллекта, обычно довольно грубое); | нормальный интеллект. |
| органов зрения (помутнение роговицы, глаукома); | Патология других органов и систем идентична изменениям у пациентов с Гурлер-подобным фенотипом мукополисахаридоза. |
| слуха (тугоухость различной степени выраженности); | |
| сердечно-сосудистой системы (недостаточность клапанов сердца, гипертрофия миокарда, нарушения сердечного ритма); | |
| бронхолёгочной системы (синусобронхопатии с образованием обильного количества слизисто-гнойного отделяемого, снижение показателей функции внешнего дыхания, апноэ). | |
Симптомы
Все пациенты, страдающие мукополисахаридозом, имеют характерные отличительные фенотипические признаки в виде развития уродливых черт лица с увеличенным языком и большой головой. Больные любым типом мукополисахаридоза имеют:
- признаки отставания в физикометрическом развитии, а именно несоответствие костного возраста паспортному,
- развитие выраженной степени сколиоза и обезображивающих контрактур крупных суставов.
- Несмотря на преимущественное поражение нервной и/или опорно-двигательной системы, при некоторых типах мукополисахаридоза отмечается повреждение внутренних органов.
- Характерными отличительными симптомами считается поражение нейронов корковых структур головного мозга, вследствие чего у ребенка развиваются признаки нарушения интеллектуальной способности различной степени интенсивности.
- При прогрессивном течении заболевания практически в 100% случаев мукополисахаридоза отмечается поражение зрительного нерва, а также роговицы одного/обоих глаз, сопровождающееся выраженным нарушением зрительной функции.
Диагностика
Диагноз МПС в течение первого года жизни устанавливается редко, поскольку первые его симптомы неспецифичны. Патогномоничным лабораторным признаком заболевания является обнаружение увеличенной экскреции гепарани дерматансульфата с мочой и подтверждение дефицита фермента в лейкоцитах или фибробластах.
Также доступно генетическое исследование, при котором обнаруживаются мутации в гене IDUA (4p16.3), что приводит к полному дефициту фермента α-L-идуронидазы. Рентгенологически выявляют преждевременное окостенение затылочно-теменного шва, выраженное удлинение турецкого седла.
Характерна деформация тел позвонков («рыбий позвонок»), искривление лучевой кости, деформации в области ядер окостенения метафизов и эпифизов длинных трубчатых костей, короткие метакарпальные кости, фаланги в форме сахарной головы при нормальном развитии ядер окостенения костей (дифференциально-диагностический признак).
Лечение
Терапия должна включать симптоматическую коррекцию нарушенных функций внутренних органов (хирургическое, ортопедическое лечение). Необходимо наблюдение офтальмологом, логопедом, психиатром.
В последние годы разработан метод трансплантации гемопоэтических стволовых клеток, который предпочтителен у пациентов младше 2,5 лет. Указанный метод способствует увеличению продолжительности и качества жизни, позволяет сохранить нейрокогнитивность.
Для всех пациентов с синдромом Гурлер для облегчения симптомов не неврологического характера рекомендуется пожизненная ферментозаместительная терапия ларонидазой.
Несмотря на лечение, пациенты часто погибают в первом десятилетии жизни от осложнений со стороны сердечной и дыхательной систем.
Мукополисахаридоз 1 типа. Лечение
Поскольку мукополисахаридоз 1 типа является генетическим расстройством, то назначаемые терапии будут содержать как лечебные так и паллиативные элементы. Фермент заместительная терапия лоранидазой может обеспечить клинически важные преимущества, такие как улучшение легочной функции и сокращение избыточных углеводов, хранящихся в органах.
Хирургическая помощь
Корректирующие операции могут быть необходимы для пациентов с мукополисахаридозом 1 типа, которые имеют контрактуры суставов, а также уродства рук и ног. Некоторым пациентам может потребоваться пересадка роговицы, если проблемы со зрением будут серьезными.