Eprex
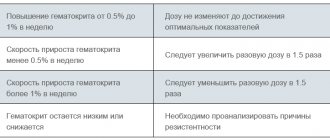
During treatment with Eprex, blood pressure should be monitored weekly and a complete blood count (including platelets, hematocrit, ferritin) should be performed. In the pre- and postoperative period, Hb should be monitored more often if the initial value was less than 140 g/l.
It must be remembered that treatment of anemia with Eprex does not replace blood transfusion, but reduces the need for its repeated use.
In patients with controlled arterial hypertension or a history of thrombotic complications, an increase in the dose of antihypertensive drugs and anticoagulants may be required, respectively.
When prescribed to patients with liver failure, a slowdown in the metabolism of epoetin and a pronounced increase in erythropoiesis are possible. The safety of the drug in this category of patients has not been established. Although the drug stimulates erythropoiesis, the possibility of Eprex affecting the growth of certain types of tumors, including bone marrow.
The possibility that a preoperative increase in Hb may predispose to the development of thrombotic complications should be considered. Before undergoing elective surgery, patients should receive adequate prophylactic antiplatelet therapy. In the pre- and postoperative period, it is not recommended to prescribe it to patients with an initial Hb of more than 150 g/l. In adult patients with chronic renal failure and clinically significant ischemic heart disease or heart failure, Hb should not exceed 100-120 g/l.
Before starting treatment, possible causes of an inadequate reaction to the drug should be excluded (deficiency of Fe, folic acid, cyanocobalamin, severe Al3+ poisoning, concomitant infections, inflammatory processes and injuries, hidden bleeding, hemolysis, bone marrow fibrosis of various etiologies) and, if necessary, adjust the treatment. Before starting treatment, iron reserves in the body should be assessed. In most patients with chronic renal failure, in cancer and HIV-infected patients, the plasma ferritin concentration decreases simultaneously with an increase in hematocrit. Ferritin concentrations must be determined throughout the course of treatment. If it is less than 100 ng/ml, replacement therapy with oral Fe preparations is recommended at a rate of 200-300 mg/day (100-200 mg/day for children). Patients donating autologous blood and in the pre- or postoperative period should also receive an adequate amount of Fe orally at a dose of 200 mg/day.
In patients with chronic renal failure, correction of anemia may result in improved appetite and increased absorption of K+ and proteins. Periodic adjustments of dialysis parameters may be required to maintain BUN, creatinine, and K+ concentrations within normal limits. In patients with chronic renal failure, it is necessary to monitor serum electrolytes.
According to available data, the use of Eprex in predialysis patients does not accelerate the progression of chronic renal failure. Due to increased hematocrit, it is often necessary to increase the dose of heparin during hemodialysis. With inadequate heparinization, blockage of the dialysis system and thrombosis of shunts are possible, especially in patients with a tendency to hypotension or with complications of an arteriovenous fistula (stenosis, aneurysm, etc.). In such patients, early revision of the shunt and timely prevention of thrombosis (for example, ASA) are recommended.
Use during pregnancy and lactation is possible only in cases where the potential benefit to the mother outweighs the potential risk to the fetus. It is not known whether the drug is excreted in breast milk. When used in women of reproductive age with anemia due to chronic renal failure, menstruation may resume. The patient should be warned about the possibility of pregnancy and the need to use reliable methods of contraception before starting therapy.
Experimental studies on rats and rabbits did not reveal a teratogenic effect when administered intravenously in doses up to 500 IU/kg body weight per day; at higher doses, a weak, statistically insignificant decrease in fertility was noted.
Chronic toxicity studies (in rats and dogs) revealed the development of subclinical fibrosis of bone marrow tissue. During the 13-week study, dogs treated subcutaneously or intravenously at doses of 80, 240, or 520 units/kg/day developed anemia without or with signs of bone marrow hypoplasia. Due to the fact that epoetin alfa is a human glycoprotein, it is recognized that these changes could be caused by the action of antibodies to epoetin alfa. Similar phenomena were observed in isolated cases when using the drug in veterinary practice and were explained by the appearance of antibodies to epoetin alfa. No carcinogenicity studies have been conducted. Does not cause gene mutations in bacteria (Ames test), chromosomal aberrations in mammalian cells, micronucleia in mice, or gene mutations in the HGRT locus.
During treatment with Eprex, until the optimal maintenance dose is established in patients with chronic renal failure, caution must be exercised when driving vehicles and engaging in other potentially hazardous activities that require increased concentration and speed of psychomotor reactions (increased risk of increased blood pressure at the beginning of therapy).
pharmachologic effect
Erythropoiesis stimulator. It is a purified glycoprotein. Affects the division and differentiation of progenitor cells. Epoetin alfa is produced by mammalian cells with an integrated gene encoding the synthesis of human erythropoietin. The biological properties of epoetin alfa are no different from human erythropoietin.
The protein fraction makes up about 58% of the molecular weight and consists of 165 amino acids. The four hydrocarbon chains are attached to the protein by three N-glycosidic bonds and one O-glycosidic bond. The molecular weight of erythropoietin is approximately 32,000-40,000 daltons.
After administration of the drug, the number of red blood cells, reticulocytes, hemoglobin level and the rate of 59Fe absorption increase. In bone marrow cell culture, it was shown that epoetin alfa selectively stimulates erythropoiesis without affecting leukopoiesis.
Epoetin alfa has minimal ability to induce antibody formation.
No carcinogenicity studies have been conducted.
Epoetin alfa does not cause mutations in bacterial genes (Ames test), chromosomal aberrations in mammalian cells, or mutations in the HGPRT locus genes.
One study found no difference in the incidence of bone marrow fibrosis between dialysis patients who received epoetin alfa for three years and similar patients who did not receive epoetin alfa.
Recommendations of the Russian Ministry of Health
The Ministry of Health of the Russian Federation has updated anti-Covid recommendations and approved the 9th version. The main changes in it concern quarantine measures and outpatient treatment of patients with Covid-19.
The 9th version of the recommendations also includes treatment regimens for coronavirus infection in a hospital setting. Actemra appears in sections that deal with severe and critical forms of the disease.
Why Actemra
French scientists were among the first to discover the effectiveness of Actemra in treating cytokine expression in coronavirus. They announced that the formidable complication was overcome by the arthritis drug Actemra (Tocilizumab).
According to doctors, the use of Actemra “significantly improves the prognosis for moderate and severe forms of coronavirus pneumonia.” In such cases, hypercytokinemia often develops - the same cytokine storm that can cause unpredictable consequences.
As a result of a cytokine storm, the body's own cells are attacked by cytokine proteins. It was this process that doctors tried to stop with Actemra, a drug intended for the treatment of rheumatoid arthritis.
The French experiment involved 65 patients receiving Actemra. Among them, there were fewer cases of connection to mechanical ventilation and a significantly lower mortality rate recorded within 2 weeks from the onset of the disease.
Contraindications
- patients with partial red cell aplasia treated with any erythropoietin;
- uncontrolled arterial hypertension;
- severe pathology of the coronary, carotid, cerebral and peripheral vessels, including recent myocardial infarction or acute cerebrovascular accident (as part of a pre-deposit blood collection program before major surgery);
- patients who, for some reason, are unable to receive adequate preventive antithrombotic therapy;
- pregnancy;
- lactation period (breastfeeding);
- hypersensitivity to the components of the drug.
Directions for use and doses
Before use, carefully inspect the solution for visible particles or discoloration. The drug should not be shaken, because this may lead to denaturation of the glycoprotein and loss of drug activity. Eprex® does not contain preservatives, so individual packaging is intended for single use.
IV administration
The duration of the injection is at least 1-5 minutes. Slower administration is preferable for patients who develop a flu-like syndrome when the drug is administered.
It is prohibited to administer the drug as an intravenous infusion or mix it with other medications.
SC introduction
The maximum volume of one subcutaneous injection should not exceed 1 ml; if it is necessary to administer large volumes, several injection points should be used. The drug is injected under the skin of the shoulder, thigh, and anterior abdominal wall.
When changing the route of administration, the drug is administered at the same dose, then the dose is adjusted if necessary (to achieve the same therapeutic effect with subcutaneous administration, a dose of 20-30% less than with intravenous administration is required.
Scientific data
The experiment involved 5,776 patients from the American clinic in New York, Nortwell Heaith. All of them had identical and strictly defined initial parameters, but received different treatments.
The patients were divided into 6 groups, the largest of which consisted of 3076 people and received standard therapy that did not include immunomodulators.
The second group of 1383 people received hormonal treatment.
The third group of patients (454 people) received Actemra, or Tocilizumab, which is the name of its active substance.
In the fourth group, the treatment regimen included steroid hormones + Actemra, in the fifth (73 people) - only Actemra, and patients from the 6th group (57 people) were treated with Anakinra.
The subject of the study was in-hospital mortality.
The lowest mortality was found in the group whose members received steroids in combination with Actemra. It was lower than in groups receiving standard therapy, steroid monotherapy, and hormones in combination with Anakinra.
Pharmacological properties of Eprex
By nature, Eprex is a glycoprotein epoetin alpha obtained by genetic engineering. It belongs to the group of pharmacological agents that stimulate the formation of red blood cells - erythrocytes, and does not differ in properties from human erythropoietin.
The mechanism of action of the drug is that it stimulates the division and differentiation of red blood cell precursors. This effect is selective in nature, without affecting, for example, the formation of white blood cells - leukocytes. Research has proven that Eprex has a slight ability to induce the formation of antibodies to it.
There is no evidence that Eprex has teratogenic or carcinogenic properties.
Indications for use
- anemia associated with chronic renal failure in adults and children (including in patients on hemo- or peritoneal dialysis);
- anemia in cancer patients with non-myeloid tumors (for prevention and treatment);
- anemia in HIV-infected patients receiving zidovudine therapy with endogenous erythropoietin levels less than 500 IU/ml;
- as part of a predeposit program before major surgery in patients with a hematocrit level of 33-39%, to facilitate the collection of autologous blood and reduce the risk associated with the use of allogeneic blood transfusions if the expected need for transfusion exceeds the amount that can be obtained by autologous collection without the use of epoetin alfa;
- before undergoing major surgery with an expected blood loss of 900-1800 ml (2-4 units) in adult patients with no or mild to moderate anemia (hemoglobin level 100-130 g/L) to reduce the need for allogeneic blood transfusions and facilitate recovery erythropoiesis.
Properties and characteristics
Actemra (tocilizumab) is an immunosuppressant that inhibits the IL-6 cytokine subclass of IgG antibodies. It is prescribed in the complex therapy of rheumatoid arthritis (RA) with moderate or high activity.
Tocilizumab is used in children from 2 years of age with active forms of polyarticular and juvenile idiopathic RA.
The effectiveness of the drug, used both as monotherapy and in combination with Methotrexate and other basic drugs, does not depend on the presence (or absence) of rheumatoid factor, as well as age, gender, race, the number of previous therapeutic courses or the stage of the disease.
The response to treatment appears quickly, already in the second week of use, then increases and persists for 3 years, which is confirmed by open-label extended studies.