Erythropoietin is a drug (solution) that belongs to the pharmacological group of antianemic drugs. Important features of the medicine from the instructions for use:
How to dissolve vascular plaques, normalize blood circulation, blood pressure and forget the way to the pharmacy
- Sold only with a doctor's prescription
- During pregnancy: with caution
- When breastfeeding: with caution
- For liver dysfunction: with caution
- If renal function is impaired: with caution
Release form and composition
Erythropoietin is available in the form of a solution for intravenous (IV) and subcutaneous (SC) administration: colorless transparent liquid [500 IU (international unit) or 2000 IU in ampoules of 1 ml, 5 ampoules in blister packs, in cardboard pack 1 or 2 packages].
1 ml of solution contains:
- active ingredient: epoetin beta (recombinant human erythropoietin) 500 IU or 2000 IU;
- auxiliary components: isotonic citrate buffer (sodium chloride, sodium citrate, water for injection, citric acid), albumin solution 10%.
Erythropoietin
Active substance:
Epoetin beta*
Pharmgroup:
Hematopoiesis stimulants
Average price in pharmacies
| Name | Manufacturer | average price |
| Erythropoietin 2000IU/ml 1ml n10 amp solution i.v. s.c. | OBOLENSKOYE | 7269.00 |
Analogs for the active substance:Vero-Epoetin Recormon Epostim Epoetin Human erythropoietin recombinant |
Pharmacological properties
Pharmacodynamics
Erythropoietin is a drug for increasing hemoglobin and hematocrit levels in the body, improving heart function and blood supply to tissues.
Active ingredient: epoetin beta; being a glycoprotein, its biological and immunological properties and composition are identical to natural human erythropoietin. Epoetin beta has the property of specifically stimulating the process of hematopoiesis in the body, activating mitosis and maturation of red blood cells from the precursor cells of the erythrocyte series. The synthesis of recombinant epoetin beta occurs in mammalian cells into which the gene with the code for human erythropoietin is inserted.
The most pronounced therapeutic effect of Erythropoietin occurs in anemia caused by chronic renal failure.
Long-term use of the drug in rare cases can cause the formation of antibodies that neutralize the effect of epoetin beta and contribute to the development of partial red cell aplasia.
Pharmacokinetics
The bioavailability of Erythropoietin with subcutaneous administration is 25–40%.
The half-life for intravenous administration is from 4 to 12 hours, for subcutaneous administration it is 13–28 hours.
Overdose of Epovitan™ recombinant human erythropoietin, symptoms and treatment
An overdose of the drug does not cause toxic effects. But if the main indicators (hemoglobin, hematocrit) are not adjusted during treatment with recombinant human erythropoietin, then their optimal and maximum permissible values are exceeded. Exceeding the dose of erythropoietin can cause an increase in blood pressure and contribute to the development of hypertensive encephalopathy (in severe cases with convulsions). In the absence of correction for the hematocrit index, polycythemia may develop, to reduce the severity of which phlebotomy is prescribed (to normalize the hematocrit index).
Indications for use
According to the instructions, Erythropoietin is indicated for the prevention and treatment of anemia in the following diseases and conditions:
- treatment of anemia caused by chronic renal failure, including in patients on dialysis;
- prevention and treatment of anemia in adults with solid tumors receiving platinum chemotherapy at a cyclic dose that may cause anemia;
- treatment of anemia in adults with a relative deficiency of endogenous erythropoietin receiving antitumor therapy for myeloma, low-grade non-Hodgkin's lymphoma, chronic lymphocytic leukemia;
- prevention of anemia in premature newborns born before the 34th week of pregnancy with a body weight of 0.75–1.5 kg.
In addition, Erythropoietin is used to increase the volume of donor blood intended for autotransfusion.
Contraindications
- partial red cell aplasia due to previous therapy with epoetin beta;
- inability to carry out adequate anticoagulant therapy;
- uncontrolled arterial hypertension;
- period within a month after myocardial infarction;
- unstable angina;
- increased risk of deep vein thrombosis, thromboembolism when collecting blood before surgery;
- porphyria;
- hypersensitivity to the components of the drug.
Caution should be exercised when using Erythropoietin in patients with moderate anemia without iron deficiency, sickle cell anemia, refractory anemia, malignant neoplasms, a history of thrombosis, thrombocytosis, chronic liver failure, epilepsy, nephrosclerosis, autotransfusion in patients with a body weight of up to 50 kg.
During pregnancy and lactation, the use of Erythropoietin is indicated only in exceptional cases, if, in the opinion of the doctor, the expected effect exceeds the possible threat to the mother and fetus/child.
Instructions for use of Erythropoietin: method and dosage
Erythropoietin solution is used by subcutaneous and intravenous administration. S.C. administration is more preferable.
When administered intravenously, the dose of the solution should be administered within 2 minutes.
For hemodialysis patients, Erythropoietin is administered at the end of the dialysis session through an arteriovenous shunt.
The doctor sets the dosage, treatment regimen and duration of therapy individually, taking into account the nature of the disease and the clinical indications of the patient.
Recommended dosing regimen of Erythropoietin for adults in the treatment of anemia in chronic renal failure:
- initial therapy (correction stage): subcutaneous administration - at the rate of 20 IU per 1 kg of patient’s body weight 3 times a week. In the absence of a sufficient increase in hematocrit (less than 0.5% per week), an increase in the single dose by 20 IU per 1 kg of body weight every 4 weeks is indicated. The weekly dose of the drug can be administered once or evenly distributed over daily administrations. IV administration - at the rate of 40 IU per 1 kg of body weight 3 times a week. If the hematocrit level does not increase sufficiently after 4 weeks of therapy, the single dose can be increased to 80 IU per 1 kg of weight. If necessary, the single dose can be further increased once every 4 weeks by 20 IU per 1 kg of weight. The maximum weekly dose for any route of administration should not exceed 720 IU per 1 kg of patient body weight;
- maintenance therapy: initial dose - maintaining hematocrit at 30–35% is achieved by administering a dose of ½ of the previous injection. Next, the dosage is selected individually, adjusting it once every 1–2 weeks.
The dose prescribed for the treatment of children is made taking into account the age of the child; as a rule, the older the child is, the lower the dose is required. It is advisable to start treatment with the recommended regimen.
The duration of treatment is lifelong; therapy can be interrupted at any time.
Recommended dosage of Erythropoietin:
- prevention of anemia in premature newborns: subcutaneously - 250 IU per 1 kg of body weight 3 times a week. It is necessary to start administering the drug from the third day of the child’s life and continue for 6 weeks;
- prevention and treatment of anemia in patients with solid tumors receiving chemotherapy with platinum drugs (administration of the drug is indicated only when the hemoglobin level before chemotherapy is not higher than 130 g/l): initial dose - 450 IU per 1 kg of body weight per week. If the hemoglobin level does not increase sufficiently after 4 weeks of therapy, the dose of the drug is doubled. The duration of treatment after the end of chemotherapy is no more than 3 weeks. If the hemoglobin level decreases by more than 10 g/l during the first cycle of chemotherapy, further use of the drug may not be advisable. The hemoglobin level should not be allowed to increase by more than 20 g/l within 4 weeks or exceed 140 g/l. If within 4 weeks the hemoglobin level has risen by more than 20 g/l, the dose of the drug must be reduced by 50%. When the hemoglobin concentration level is above 140 g/l, temporary discontinuation of the drug is required. After the hemoglobin level in the blood reaches less than 120 g/l, treatment should be resumed at a dose corresponding to ½ of the previous weekly dose;
- treatment of anemia due to deficiency of endogenous erythropoietin in myeloma, low-grade non-Hodgkin's lymphoma or chronic lymphocytic leukemia: initial dose - subcutaneous injection at the rate of 450 IU per 1 kg of body weight per week, the dose can be divided into 3 or 7 injections. If the hemoglobin level increases by 10 g/l after 4 weeks of therapy, treatment should be continued at the same dose. If during the specified period the hemoglobin level increases by less than 10 g/l, then the weekly dose can be increased to 900 IU per 1 kg of body weight. If, after 8 weeks of using Erythropoietin, the hemoglobin concentration level has not increased even by 10 g/l, further therapy is inappropriate and should be discontinued. It should be taken into account that the response to epoetin beta therapy in chronic lymphocytic leukemia occurs 2 weeks later than in other forms of neoplasms. After completion of chemotherapy, treatment should be continued for 4 weeks. The maximum weekly dose is no more than 900 IU per 1 kg of body weight. If the hemoglobin level increases by more than 20 g/l over 4 weeks of treatment, then treatment should be continued at a dose corresponding to ½ of the previous dose. If the level of hemoglobin concentration in the blood plasma is above 140 g/l, then treatment is temporarily stopped. Erythropoietin use can be resumed when hemoglobin levels are below 130 g/l, provided that the anemia is most likely caused by a lack of epoetin beta. The dose is prescribed 2 times less than the previous weekly dose.
Patients are prepared for autohemotransfusion by intravenous or subcutaneous administration of the drug 2 times a week for 4 weeks. The dose of Erythropoietin is determined for each patient individually, since it depends on the expected volume of donated blood taken and on the patient’s endogenous erythrocyte reserve.
If the hematocrit is above 33% and blood can be drawn without prior preparation, then epoetin beta is administered at the end of the procedure. The hematocrit throughout the entire course of therapy should not exceed 48%. The maximum weekly dose for intravenous administration should not exceed 1600 IU per 1 kg of patient body weight, for subcutaneous administration - 1200 IU per 1 kg.
Instructions:
Clinical and pharmacological group
19.002 (Erythropoiesis stimulator)
Release form, composition and packaging
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 amp. | |
| epoetin alfa (recombinant human erythropoietin) | 1000 IU |
| -«- | 2000 IU |
| -«- | 4000 IU |
| -«- | 10,000 IU |
Excipients: albumin (solution), sodium citrate pentasesquihydrate or sodium citrate dihydrate, sodium chloride, citric acid monohydrate, water for injection.
1 ml - colorless glass ampoules (5) - contour cell packaging (1) - cardboard packs. 1 ml - colorless glass ampoules (5) - contour cell packaging (2) - cardboard packs.
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 syringe | |
| epoetin alfa (recombinant human erythropoietin) | 1000 IU |
| -«- | 2000 IU |
Excipients: albumin (solution), sodium citrate dihydrate or sodium citrate pentasesquihydrate, sodium chloride, citric acid monohydrate, water for injection.
0.3 ml - syringes (3) - blister packs (1) - cardboard packs. 0.3 ml - syringes (3) with a needle protection device - blister packs (1) - cardboard packs. 0.3 ml - syringes (3) - blister packs contour (2) - cardboard packs. 0.3 ml - syringes (3) with a needle protection device - contour cell packaging (2) - cardboard packs.
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 syringe | |
| epoetin alfa (recombinant human erythropoietin) | 2000 IU |
Excipients: albumin (solution), sodium citrate dihydrate or sodium citrate pentasesquihydrate, sodium chloride, citric acid monohydrate, water for injection.
0.5 ml - syringes (3) - blister packs (1) - cardboard packs. 0.5 ml - syringes (3) with a needle protection device - blister packs (1) - cardboard packs. 0.5 ml - syringes (3) - blister packs contour (2) - cardboard packs. 0.5 ml - syringes (3) with a needle protection device - contour cell packages (2) - cardboard packs.
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 syringe | |
| epoetin alfa (recombinant human erythropoietin) | 4000 IU |
Excipients: albumin (solution), sodium citrate dihydrate or sodium citrate pentasesquihydrate, sodium chloride, citric acid monohydrate, water for injection.
0.4 ml - syringes (3) - blister packs (1) - cardboard packs. 0.4 ml - syringes (3) with a needle protection device - blister packs (1) - cardboard packs. 0.4 ml - syringes (3) - blister packs contour (2) - cardboard packs. 0.4 ml - syringes (3) with a needle protection device - contour cell packaging (2) - cardboard packs.
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 syringe | |
| epoetin alfa (recombinant human erythropoietin) | 10,000 IU |
Excipients: albumin (solution), sodium citrate dihydrate or sodium citrate pentasesquihydrate, sodium chloride, citric acid monohydrate, water for injection.
0.6 ml - syringes (3) - blister packs (1) - cardboard packs. 0.6 ml - syringes (3) with a needle protection device - blister packs (1) - cardboard packs. 0.6 ml - syringes (3) - blister packs contour (2) - cardboard packs. 0.6 ml - syringes (3) with a needle protection device - contour cell packaging (2) - cardboard packs.
The solution for intravenous and subcutaneous administration is transparent, colorless.
| 1 syringe | |
| epoetin alfa (recombinant human erythropoietin) | 10,000 IU |
Excipients: albumin (solution), sodium citrate dihydrate or sodium citrate pentasesquihydrate, sodium chloride, citric acid monohydrate, water for injection.
1 ml - syringes (3) - blister packs (1) - cardboard packs. 1 ml - syringes (3) with a needle protection device - blister packs (1) - cardboard packs. 1 ml - syringes (3) - blister packs contour (2) - cardboard packs. 1 ml - syringes (3) with a needle protection device - contour cell packaging (2) - cardboard packs.
pharmachologic effect
Erythropoiesis stimulator. Epoetin alfa is a glycoprotein that specifically stimulates erythropoiesis, activates mitosis and maturation of erythrocytes from erythrocyte precursor cells. Recombinant epoetin alfa is synthesized in mammalian cells into which the gene encoding human erythropoietin is integrated. In its composition, biological and immunological properties, epoetin alfa is identical to natural human erythropoietin.
The administration of epoetin alfa leads to an increase in hemoglobin and hematocrit levels, improving blood supply to tissues and heart function. The most pronounced effect of the use of epoetin alfa is observed in anemia caused by chronic renal failure.
In very rare cases, with long-term use of erythropoietin for the treatment of anemic conditions, the formation of neutralizing antibodies to erythropoietin with or without the development of partial red cell aplasia may be observed.
Pharmacokinetics
Suction and distribution
With subcutaneous administration of epoetin alfa, its concentration in the blood increases slowly and reaches a maximum in the period from 12 to 18 hours after administration, T1/2 is 16-24 hours. The bioavailability of epoetin alfa with subcutaneous administration is 25-40%. Does not accumulate.
Removal
With intravenous administration of epoetin alfa in healthy individuals and patients with uremia, T1/2 is 5-6 hours.
Dosage
Treatment of anemia in patients with chronic renal failure
Adult patients undergoing hemodialysis
Eralfon® is administered subcutaneously or intravenously at the end of the dialysis session. When changing the method of administration, the drug is administered at the same dose, then the dose is adjusted if necessary (with the subcutaneous route of administration of the drug, to achieve the same therapeutic effect, a dose of 20-30% less is required than with intravenous administration). Treatment with the drug includes two stages:
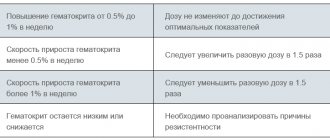
1. Correction stage: with subcutaneous administration of the drug, the initial single dose is 30 IU/kg 3 times a week. When administered intravenously, the initial single dose is 50 IU/kg 3 times a week. The correction period lasts until the optimal hemoglobin content (100-120 g/l in adults and 95-110 g/l in children) and hematocrit (30-35%) is achieved. These indicators must be monitored weekly.
The following situations are possible:
1. Hematocrit increases from 0.5 to 1.0% per week. In this case, the dose is not changed until optimal values are achieved.
2. The rate of increase in hematocrit is less than 0.5% per week. In this case, it is necessary to increase the single dose by 1.5 times.
3. Growth rate of more than 1.0% per week. In this case, it is necessary to reduce the single dose of the drug by 1.5 times.
4. Hematocrit remains low or decreases. It is necessary to analyze the causes of resistance before increasing the dose of the drug.
The effectiveness of therapy depends on a correctly selected individual treatment regimen.
2. Maintenance therapy stage: to maintain the hematocrit at a level of 30-35%, the dose of the drug used at the correction stage should be reduced by 1.5 times. Then the maintenance dose of the drug is selected individually, taking into account the dynamics of hematocrit and hemoglobin.
In children undergoing hemodialysis, the initial single dose is 50 IU/kg 3 times a week subcutaneously or intravenously. If necessary, the single dose is increased once every 4 weeks by 25 IU/kg until the optimal hemoglobin concentration is achieved. Maintenance single dose in children weighing less than 10 kg - 75-150 IU/kg (average 100 IU/kg), 10-30 kg - 60-150 IU/kg (average 75 IU/kg), more than 30 kg - 30-100 IU/kg (average 33 IU/kg) 3 times a week.
For adult predialysis patients, the initial dose is administered subcutaneously or intravenously 3 times 50 IU/kg per week. If necessary, the single dose is increased once every 4 weeks by 25 IU/kg until the optimal hemoglobin concentration is achieved. Maintenance dose - 17-33 IU/kg 3 times a week subcutaneously or intravenously.
Prevention and treatment of anemia in patients with solid tumors
Before starting treatment, it is recommended to determine the concentration of endogenous erythropoietin. When the concentration of erythropoietin in the blood serum is less than 200 IU/ml, the initial single dose of the drug is 150 IU/kg 3 times a week subcutaneously for the intravenous route of administration.
If after 4 weeks of treatment the hemoglobin level has increased and is at least 10 g/l, or the reticulocyte count has increased by more than 40,000 cells/μl above the initial amount, then the dose of Eralfon remains the same (150 IU/kg 3 times a week).
If after 4 weeks of treatment the increase in hemoglobin level is less than 10 g/l, and the increase in the number of reticulocytes is less than 40,000 cells/μl compared to the initial amount, then over the next 4 weeks the dose is increased to 300 IU/kg 3 times week.
If after an additional 4 weeks of treatment at a dose of Eralfon 300 IU/kg, the hemoglobin level has increased and is at least 10 g/l, or the number of reticulocytes has increased by more than 40,000 cells/μl, then maintain the existing dose of Eralfon (300 IU/kg 3 times in Week).
If after 4 weeks of treatment at a dose of 300 IU/kg body weight, the hemoglobin level increases by less than 10 g/l and the increase in the reticulocyte count is less than 40,000 cells/μl compared with the initial amount, treatment should be discontinued.
If the hemoglobin level increases by more than 20 g/l within a month, the dose of the drug must be reduced by 25%.
If the hemoglobin content exceeds 140 g/l, it is necessary to suspend treatment until the hemoglobin concentration decreases below 120 g/l and then continue administration of Eralfon at a dose 25% lower than the original one.
Eralfon therapy should continue for 1 month after completion of chemotherapy.
Serum ferritin levels (or serum iron levels) should be determined in all patients before and during treatment with Eralfon. If necessary, additional iron supplementation is prescribed.
Prevention and treatment of anemia in patients with HIV infection
It is recommended to determine the initial content of endogenous erythropoietin in the blood serum before starting treatment with Eralfon®. Studies have shown that if the erythropoietin content is more than 500 IU/ml, the effect of drug therapy is unlikely.
1. Correction stage: the drug is prescribed in a single dose of 100 IU/kg 3 times a week subcutaneously or intravenously for 8 weeks. If after 8 weeks of therapy it has not been possible to achieve a satisfactory effect (for example, to reduce the need for blood transfusions or to achieve an increase in hemoglobin levels), the dose can be gradually increased (no more than once every 4 weeks) by 50-100 IU/kg 3 times week. If it was not possible to achieve a satisfactory effect from therapy with Eralfon® at a single dose of 300 IU/kg 3 times a week, then a response to further therapy at higher doses is unlikely.
2. Maintenance therapy phase: after achieving a satisfactory effect in the anemia correction phase, the maintenance dose should provide a hematocrit within 30-35% and depending on changes in the dose of zidovudine, the presence of concomitant infectious or inflammatory diseases. If hematocrit (more than 40%), the drug should be discontinued until the hematocrit decreases to 36%. When restarting therapy, the dose of epoetin alfa should be reduced by 25%, followed by adjustment to maintain the required hematocrit. Serum ferritin should be determined in all patients before and during treatment treatment with the drug. If necessary, an additional iron supplement is prescribed.
Prevention and treatment of anemia in patients with multiple myeloma, low-grade non-Hodgkin lymphomas and chronic lymphocytic leukemia
In these patients, the advisability of treatment with epoetin alfa is determined by the inadequate synthesis of endogenous erythropoietin against the background of the development of anemia. When the hemoglobin level is below 100 g/l and the content of erythropoietin in the blood serum is below 100 IU/ml, Eralfon® is administered subcutaneously in a starting single dose of 100 IU/kg 3 times a week. Laboratory monitoring of hemodynamic parameters is carried out weekly. If necessary, the dose of the drug is adjusted upward or downward every 3-4 weeks. If, upon reaching a weekly dose of 600 IU/kg, no increase in hemoglobin concentration is observed, further use of epoetin alfa should be discontinued as ineffective.
Prevention and treatment of anemia in patients with rheumatoid arthritis
In patients with rheumatoid arthritis, suppression of the synthesis of endogenous erythropoietin is observed under the influence of increased concentrations of anti-inflammatory cytokines. Treatment of anemia in these patients is carried out with the drug with subcutaneous administration at a dose of 50-75 IU/kg 3 times a week. If the hemoglobin level increases by less than 10 g/l after 4 weeks of treatment, the dose of the drug is increased to 150-200 IU/kg 3 times a week. Further increase in dose seems inappropriate.
Treatment and prevention of anemia in premature infants born with low birth weight
Eralfon® is administered subcutaneously at a dose of 200 IU/kg 3 times a week, starting from the 6th day of life, until the target levels of hemoglobin and hematocrit are achieved, but not more than 6 weeks.
Adult patients participating in a pre-surgical autologous blood collection program
It is recommended to use the intravenous route of administration of the drug. Epoetin alfa should be administered at the end of the blood collection procedure. Before prescribing Eralfon, all contraindications to autologous blood collection should be taken into account. Before surgery, Eralfon® should be prescribed 2 times a week for three weeks. At each doctor's visit, a portion of blood is taken from the patient (if hematocrit ≥33% and/or hemoglobin level ≥110 g/L) and stored for autologous transfusion. The recommended dose of Eralfon® is 600 IU/kg body weight IV 2 times a week.
Serum ferritin levels (or serum iron levels) should be determined in all patients before and during treatment with Eralfon®. If necessary, additional iron intake is prescribed.
If anemia is present, its cause should be established before starting Eralfon therapy. It is necessary to ensure adequate iron intake into the body as soon as possible by prescribing an iron supplement at a dose of 200 mg/day (based on ferrous iron) and maintain iron intake at this level throughout the entire course of therapy.
Patients in the pre- and postoperative period who are not participating in the autologous blood collection program
Eralfon® is recommended to be administered subcutaneously at a dose of 600 IU/kg body weight per week for three weeks preceding surgery (21, 14 and 7 days before surgery) and on the day of surgery. If necessary, when for medical reasons it is necessary to shorten the preoperative period, Eralfon® can be prescribed daily at a dose of 300 IU/kg body weight for 10 days before surgery, on the day of surgery and for 4 days after surgery. If the hemoglobin level in the preoperative period reaches 150 g/l or higher, the use of epoetin should be discontinued. Before initiating therapy with epoetin alfa, it is necessary to ensure that patients do not have iron deficiency.
All patients should receive an adequate amount of iron (orally 200 mg/day based on ferrous iron) throughout the entire course of treatment. If possible, additional iron intake should be provided before starting therapy with Eralfon® to ensure an adequate iron depot in the patient's body.
Rules for using a syringe
- with an automatic needle protection device: you should carefully inspect the filled glass syringe with a protective device, remove the protective cap from the needle, and inject according to the standard procedure. You should press the rod with your thumb and hold it until the entire dose of the drug has been administered. The safety device is not activated until the entire dose of the drug has been administered. Next, remove the needle, release the stem, and allow the guard to move forward until the needle is completely protected and locked in place.
Avoid contact with the clamps during syringe preparation. The device is activated by pressing the rod down to the clamps.
Once the injection is complete, the needle and syringe will move back into the safety device.
- with a non-automatic needle guard: the injection should be carried out according to the standard procedure. When injecting, fingers should be kept on the safety guard to prevent premature activation of the safety device. After the injection, the protective device should be moved along the needle. An audible click will indicate the correct action. During the entire procedure, your fingers should be behind the needle.
Overdose
Symptoms: increased side effects.
Treatment: symptomatic therapy is carried out, with high hemoglobin levels - bloodletting.
Drug interactions
Reduces the concentration of cyclosporine due to an increase in the degree of its binding to red blood cells (it may be necessary to adjust the dose of cyclosporine).
Pharmaceutically incompatible with solutions of other drugs.
Use during pregnancy and lactation
Since there is insufficient experience with the use of erythropoietin during pregnancy and lactation in humans, epoetin alfa should be prescribed during pregnancy only if the expected benefits to the mother from its use exceed the possible risk to the fetus.
It is not known whether epoetin alfa is excreted in breast milk, therefore breastfeeding should be discontinued during treatment with Eralfon®.
When used in women of reproductive age with anemia due to chronic renal failure, menstruation may resume. The patient should be warned about the possibility of pregnancy and the need to use reliable methods of contraception before starting therapy.
Experimental studies on rats and rabbits did not reveal a teratogenic effect when administered intravenously in doses up to 500 IU/kg body weight/day; at higher doses, a weak, statistically insignificant decrease in fertility was noted.
Side effects
Flu-like symptoms: at the beginning of treatment - dizziness, drowsiness, fever, headache, myalgia, arthralgia.
From the cardiovascular system: dose-dependent increase in blood pressure, worsening of arterial hypertension (most often in patients with chronic renal failure); in some cases - a hypertensive crisis, a sharp increase in blood pressure with symptoms of encephalopathy (headache, confusion) and generalized tonic-clonic convulsions.
From the hematopoietic system: thrombocytosis; in some cases - thrombosis of a shunt or arteriovenous fistula (in patients on hemodialysis with a tendency to arterial hypotension or with an aneurysm, stenosis), aplasia of the erythrocyte lineage.
Allergic reactions: skin rash (mild or moderate), eczema, urticaria, itching, angioedema.
Local reactions: hyperemia, burning, mild or moderate pain at the injection site (more often occur with subcutaneous administration).
From the laboratory parameters: decrease in serum ferritin concentration, with uremia - hyperkalemia, hyperphosphatemia.
Other: complications associated with respiratory failure or decreased blood pressure, immune reactions (induction of antibody formation), exacerbation of porphyria.
Storage conditions and periods
The drug should be stored out of the reach of children at a temperature of 2° to 8°C. Shelf life: 2 years.
Indications
- anemia in patients with chronic renal failure, incl. those on hemodialysis;
— prevention and treatment of anemia in patients with solid tumors, whose anemia was a consequence of antitumor therapy;
- prevention and treatment of anemia in HIV-infected patients caused by the use of zidovudine, with a concentration of endogenous erythropoietin less than 500 IU/ml;
- prevention and treatment of anemia in patients with multiple myeloma, low-grade non-Hodgkin's lymphoma, chronic lymphocytic leukemia;
— prevention and treatment of anemia in patients with rheumatoid arthritis;
- treatment and prevention of anemia in premature babies born with low body weight up to 1.5 kg;
- as part of a predeposit program before major surgery in patients with a hematocrit level of 33-39%, to facilitate the collection of autologous blood and reduce the risk associated with the use of allogeneic blood transfusions if the expected need for transfused blood exceeds the amount that can be obtained by autologous method collection without the use of epoetin alfa;
- before undergoing major surgery with an expected blood loss of 900-1800 ml in adult patients without anemia or with mild to moderate anemia (hemoglobin concentration 100-130 g/l) to reduce the need for allogeneic blood transfusions and facilitate the restoration of erythropoiesis.
Contraindications
- partial red cell aplasia after previous therapy with any erythropoietin;
- uncontrolled arterial hypertension;
— impossibility of carrying out adequate anticoagulant therapy;
- severe occlusive diseases of the coronary, carotid, cerebral and peripheral arteries and their consequences, including acute and recent myocardial infarction and acute cerebrovascular accident (as part of a pre-deposit blood collection program before surgery);
- hypersensitivity to the drug or its components.
The drug should be prescribed with caution for malignant neoplasms, epileptic syndrome (including if there is a history), thrombocytosis, thrombosis (history), sickle cell anemia, iron or folate deficiency conditions, porphyria, chronic liver failure.
special instructions
During treatment, it is necessary to monitor blood pressure weekly and perform a complete blood count (including platelet count), determination of hematocrit and ferritin content in the blood serum. In the pre- and postoperative period, hemoglobin concentration should be monitored more often if the initial concentration was less than 140 g/l. It must be remembered that epoetin alfa in the treatment of anemia does not replace blood transfusion, but reduces the need for its repeated use.
In patients with controlled hypertension or a history of thrombotic complications, it may be necessary to increase the dose of antihypertensive drugs and/or anticoagulants, respectively.
When prescribed to patients with liver failure, a slowdown in the metabolism of epoetin alfa and a pronounced increase in erythropoiesis are possible. The safety of the drug in this category of patients has not been established.
Although the drug stimulates erythropoiesis, the possibility of epoetin alfa affecting the growth of certain types of tumors, including bone marrow.
The possibility that a preoperative increase in hemoglobin levels may predispose to the development of thrombotic complications should be considered. Before undergoing elective surgery, patients should receive adequate prophylactic antiplatelet therapy. In the pre- and postoperative period, the drug is not recommended for use in patients with an initial hemoglobin level of more than 150 g/l.
In adult patients with chronic renal failure, clinically significant ischemic heart disease or chronic heart failure, the hemoglobin concentration should not exceed 100-120 g/l.
Before starting treatment, possible causes of an inadequate reaction to the drug should be excluded (deficiency of iron, folic acid, cyanocobalamin, severe poisoning with aluminum salts, concomitant infections, inflammatory processes and injuries, hidden bleeding, hemolysis, bone marrow fibrosis of various etiologies) and, if necessary, adjust the treatment.
Before starting treatment, iron reserves in the body should be assessed. In most patients with chronic renal failure, in cancer and HIV-infected patients, plasma ferritin concentration decreases simultaneously with an increase in hematocrit level. Ferritin concentrations must be determined throughout the course of treatment. If it is less than 100 ng/ml, oral iron replacement therapy is recommended at a rate of 200-300 mg/day (100-200 mg/day for children). In premature infants, oral iron therapy at a dose of 2 mg/day should be prescribed as early as possible. Patients donating autologous blood and in the pre- or postoperative period should also receive adequate amounts of iron orally at a dose of 200 mg/day.
In patients with chronic renal failure, correction of anemia may result in improved appetite and increased absorption of potassium and protein. Periodic adjustments of dialysis parameters may be required to maintain blood urea, creatinine, and potassium concentrations within normal limits.
In patients with chronic renal failure, it is necessary to monitor the level of electrolytes in the blood serum.
According to available data, the use of epoetin alfa in predialysis patients does not accelerate the progression of chronic renal failure. Due to increased hematocrit, it is often necessary to increase the dose of heparin during hemodialysis. With inadequate heparinization, blockage of the dialysis system and thrombosis of the vascular access are possible, especially in patients with a tendency to hypotension or with complications of an arteriovenous fistula (including stenosis, aneurysm). In such patients, thrombosis prophylaxis is recommended.
Considering the possible more pronounced effect of the drug, its dose should not exceed the dose of recombinant erythropoietin used in the previous course of treatment. During the first two weeks, the dose is not changed and the dose/response ratio is assessed. After this, the dose can be reduced or increased.
Impact on the ability to drive vehicles and operate machinery
During the treatment period, until the optimal maintenance dose is established, patients with chronic renal failure must be careful when driving vehicles and engaging in other potentially hazardous activities that require increased concentration and speed of psychomotor reactions (increased risk of increased blood pressure at the beginning of therapy).
Use for liver dysfunction
When prescribed to patients with liver failure, a slowdown in the metabolism of epoetin alfa and a pronounced increase in erythropoiesis are possible. The safety of the drug in this category of patients has not been established.
Conditions for dispensing from pharmacies
The drug is available with a prescription.
Registration numbers
• solution for intravenous and subcutaneous administration of 10,000 IU/1 ml: syringes 3 or 6 pcs. with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 4000 IU/0.4 ml: syringes 3 or 6 pcs. . with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 10,000 IU/0.6 ml: 3 or 6 syringes PC. with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 2000 IU/0.5 ml: syringes 3 or 6 pcs. . with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 2000 IU/0.3 ml: syringes 3 or 6 pcs. . with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 1000 IU/1 ml: amp. 5 or 10 pcs. LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 10,000 IU/1 ml: amp. 5 or 10 pcs. LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 2000 IU/1 ml: amp. 5 or 10 pcs. LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 1000 IU/0.3 ml: syringes 3 or 6 pcs. with or without a needle protection device LSR-006663/08 (2015-08-08 – 0000-00-00) • solution for intravenous and subcutaneous administration 4000 IU/1 ml: amp. 5 or 10 pcs. LSR-006663/08 (2015-08-08 – 0000-00-00)
Side effects
- from the cardiovascular system: often - in patients with chronic renal failure, existing arterial hypertension increases or blood pressure (BP) increases; in some cases – hypertensive crisis;
- from the nervous system: in some cases - encephalopathy (including headaches, speech and gait disturbances, convulsions, confusion), migraine-like pain;
- from the hematopoietic organs: very rarely - thrombocytosis, thromboembolic complications;
- other: rarely - development of allergic reactions (rash, itching, urticaria), reactions at the injection site; in some cases - anaphylactoid reactions, transient flu-like symptoms (usually at the beginning of therapy) in the form of general malaise, fever, chills, headache, pain in the bones and limbs, increased levels of potassium and phosphates in the blood serum.
Side effects of the drug Epovitan™ recombinant human erythropoietin
At the beginning of treatment, adverse reactions manifest themselves in the form of symptoms such as fever, headache, lethargy, dizziness, drowsiness, myalgia and arthralgia. When using recombinant human erythropoietin, thrombocytosis, leukocytosis, and eosinophilia sometimes develop; biochemical studies indicate an increase in the level of total bilirubin and the activity of ALT, AST and LDH. Thrombotic vascular complications manifest themselves in the form of myocardial ischemia and infarction, cerebrovascular complications, transient ischemic attacks, deep vein thrombosis, arterial thrombosis, retinal vascular thrombosis, pulmonary embolism, aneurysm, and dialysis system occlusion. Blood pressure may increase. When administering the drug subcutaneously, pain, local skin hyperemia, rash, and itching may occur at the injection sites. Patients with renal failure from the very beginning of treatment with Epovitan need to monitor blood pressure levels, since a dose-dependent increase in blood pressure may develop or existing hypertension (arterial hypertension) may worsen, which may manifest itself as a hypertensive crisis, symptoms of encephalopathy, and generalized tonic-clonic seizures. Such side effects require immediate medical attention. During hemodialysis, complications from the arteriovenous fistula (stenosis, aneurysm, etc.) can lead to shunt thrombosis. Oncology patients Such patients may develop thrombotic complications. Surgical patients undergoing an autologous blood collection program and patients outside this program: thrombotic and vascular complications that can simultaneously arise against the background of concomitant cardiovascular diseases and repeated phlebotomy and orthopedic interventions.
special instructions
The first dose of Erythropoietin should be administered under the supervision of a physician, as there is a risk of developing an anaphylactoid reaction.
The goal of using the drug is to achieve a hematocrit volume of 30–35% of blood plasma, or eliminate the need for blood transfusion. The increase in hematocrit should not exceed 0.5% per week. Its content should not exceed 35%.
The use of Erythropoietin as a doping agent by healthy people can cause life-threatening complications from the cardiovascular system against the background of a sharp increase in hematocrit levels.
Treatment must be accompanied by weekly monitoring of blood pressure, general blood count, including determination of platelet levels, ferritin, and hematocrit. During the first 8 weeks of therapy, a cell count, especially platelets, is required. If the platelet count exceeds normal, treatment should be interrupted.
Periodically during treatment it is necessary to monitor the levels of potassium and phosphate in the blood serum. If hyperkalemia develops, the use of Erythropoietin should be discontinued until the potassium level in the blood normalizes.
In patients on hemodialysis, it is recommended to carefully monitor blood pressure, increase the dose of heparin in accordance with the increase in hematocrit, timely prevent thrombosis and early revision of the shunt.
When using Erythropoietin to increase the volume of donor blood intended for autotransfusion, the advantages of epoetin beta should first be compared with the increased risk of thromboembolism associated with its use. Therefore, for patients with moderate anemia with a hemoglobin concentration of 100–130 g/l or a hematocrit of 30–39% (without iron deficiency), the drug is recommended to be prescribed only if it is not possible to obtain a sufficient amount of canned blood for planned extensive surgery in the required volume. For women it should be more than 4 units, for men – more than 5 units.
In most cases, against the background of an increase in hematocrit, a decrease in serum ferritin levels occurs, so simultaneous administration of iron supplements in the required doses may be required.
In women of reproductive age, the use of Erythropoietin may restore menstruation. Therefore, when prescribing the drug, the doctor should warn about the possibility of pregnancy and recommend the use of reliable contraceptives.
Since Erythropoietin may have a more pronounced effect in subsequent courses of therapy, when resuming therapy, its dose should not exceed the dose of the previous course of treatment. It should not be changed during the first two weeks of therapy, then it is adjusted by assessing the dose-response relationship.
Impact on the ability to drive vehicles and complex mechanisms
Due to the increased risk of increased blood pressure at the beginning of therapy, patients with uremia should not perform potentially hazardous types of work that require increased attention and a high speed of psychomotor reactions until the optimal maintenance dose is established.
Erythropoietin in sports[edit | edit code]
The history of the use of recombinant erythropoietin in sports (abbreviations generally accepted in the scientific literature are rHuEPO, r-HuEPO, rhu-EPO, rEPO) began in 1977, when purified erythropoietin was first isolated from human urine. The introduction and fight against erythropoietin in sports and competitions as a prohibited drug went through the following stages:
- 1985 - EPO gene cloned;
- 1987 - Recombinant erythropoietin became available for the first time in Europe;
- 1987—1990 - several deaths among Dutch and Belgian cyclists have been linked to the use of EPO;
- 1988 - The International Ski Federation includes erythropoietin in the list of doping agents;
- 1989 - The FDA (Food and Drug Administration is a government agency that controls the production and distribution of drugs in the country) allows the production of recombinant EPO;
- 1990 - the use of erythropoietin is prohibited by the IOC;
- 1993-1994 — The IAAF, with the active participation of Professor M. Donike, is introducing a blood sampling procedure at eight World Cup competitions;
- 1997 - The International Cycling Union and the International Ski Federation approve random blood testing before competition, setting maximum permissible levels of hematocrit and hemoglobin. Although exceeding the established indicators is not grounds for disqualification, this procedure is aimed at protecting the athlete’s body from possible complications associated with increased hemoglobin and hematocrit;
- 1998 - exposure of cases of the use of erythropoietin in sports, at the Tour de France cycling race, is widely covered by the media;
- 1999 - Research was intensified to develop a reliable method for detecting EPO for the Sydney Olympics.
Because natural and recombinant erythropoietin have nearly identical amino acid structures, recombinant erythropoietin is extremely difficult to distinguish from its physiological counterpart.
To stimulate the secretion of its own erythropoietin, xenon inhalations are actively used in Russia. At the 2014 Sochi Olympics, many Russian athletes received xenon inhalations before the start of the competition. This method has been banned by the Anti-Doping Agency since May 2014.
Doping control[edit | edit code]
The modern arsenal of methods designed to determine erythropoietin includes direct and indirect approaches. The direct method is based on the identification of those minor differences that were found in the study of natural endogenous erythropoietin and EPO obtained by genetic engineering. In particular, some researchers have tried to exploit the differences in electrical charge distribution that have been established for these two types of EPO molecules. Based on these differences, attempts have been made to separate the two types of molecules using the method of capillary electrophoresis. And although such a separation is possible in principle, this requires large volumes of urine (up to 1 liter, which, for obvious reasons, is unacceptable for practice).
Preference is given to indirect methods that require only small volumes of blood or urine samples. Examples of an indirect method for detecting EPO are the following:
- deviations from the normal level of content in the biological environment of the sample. This fact means that the established excess of EPO levels must be distinguished from variations of a physiological or pathological nature. However, the use of this criterion is possible only if the range of fluctuations in the indicator is quite narrow compared to the values that are detected after exogenous administration of the drug. The latter is only possible when using blood as a sample for a doping test;
- registration of biochemical parameters, the value of which depends on the concentration of erythropoietin. This approach could be based on measuring serum levels of soluble transferrin receptor (sTfR), the level of which increases after administration of recombinant EPO. However, this indicator undergoes similar changes after training in mid-mountain conditions;
- determination of fibrin and fibrinogen breakdown products in urine after EPO administration.
At present, it is practically impossible to reliably identify cases of exogenous administration of erythropoietin into the body. Therefore, changes in physiological blood parameters that are detected after EPO administration are used for monitoring. Thus, the International Cycling Union uses the criterion of the maximum hematocrit value (50% for men). As such a criterion, the International Ski Federation has established the maximum permissible values of hemoglobin (165 g/l for women and 185 g/l for men), as well as a reticulocyte level of no more than 0.2%. In case of exceeding the specified limit values established during the control procedure before the competition, the corresponding athlete is suspended from participation in the competition in order to protect health. However, both hemoglobin and hematocrit are indicators that are influenced by many factors. In particular, both of these indicators can change significantly even after one moderate-volume endurance session. In addition, these indicators are characterized by significant individual variability. Therefore, simply exceeding the hematocrit value by more than 50% cannot serve as evidence of abuse of erythropoietin in sports.
To improve control over the use of erythropoietin preparations as doping, WADA introduced a modality for maintaining an athlete’s blood passport. The blood passport is one of WADA’s developments, aimed primarily at identifying erythropoietin and its analogues. With its help, a single computer hematological profile of each athlete is formed using 30 different indicators, for starters - in those sports where endurance is required. 10 countries have already joined the implementation and refinement of the blood certification program, including Sweden, Norway, Canada and Germany. The Russian Anti-Doping Agency approves of this initiative, but intends to implement it after finalizing all medical and legal aspects.
To carry out tests included in an athlete's blood passport, WADA recommends the use of equipment from Sysmex (Japan) or a subsidiary of ERMA. This brand of fully automatic hematology analyzers of the latest generation has won the maximum confidence index in the accuracy of assessing blood parameters.
During intensive training sessions and professional sports, it is necessary to constantly conduct a hematological analysis to determine the number of red blood cells and their parameters (volume, hemoglobin saturation), hemoglobin level and hematocrit. The hematocrit should not be allowed to rise above 50% - this leads to thickening of the blood, which, in turn, is fraught with deterioration of blood microcirculation in the muscles and internal organs, increasing the risk of developing thrombosis (the tendency to thrombophilia can be assessed by the D-dimer marker). In addition, complete monitoring of iron metabolism is necessary (iron concentration in serum, total and unsaturated iron-binding capacity, percentage of iron saturation, transferrin, ferritin, C-reactive protein) and determination of the level of folic acid and vitamin B12 in the blood. All these compounds are necessary for proper erythropoiesis and their deficiency should not be allowed during sports. In addition to the above tests, it is necessary to monitor the level of erythropoietin itself.
Source: