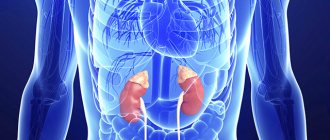
Adrenal pheochromocytoma is a hormonally active benign tumor of the adrenal glands that produces large amounts of catecholamines. Adrenal catecholamines include adrenaline and norepinephrine. Norepinephrine plays the role of a neurotransmitter; adrenaline is the main hormone of the medulla of the gland. Pheochromocytoma can develop in the medulla of the gland, or can be localized outside the adrenal glands. A malignant form of adrenal tumor is pheochromoblastoma. Pheochromocytoma is most often diagnosed between the ages of 20 and 50 years, the majority of patients are women.
One of the symptoms of the disease is the development of hypertension, which is difficult to treat. If you experience symptoms of persistent hypertension, constant anxiety, rapid heartbeat, chest pain, or unreasonable fear, you should seek help from a doctor. The Yusupov Hospital sees general practitioners, oncologists, surgeons and other specialists. The hospital is equipped with innovative equipment from leading medical equipment manufacturers in the world. Patients will be able to undergo various types of studies, including contrast, studies of hormonal levels, the condition of the abdominal organs, kidneys, and adrenal glands in the oncology department of the hospital.
Causes of pheochromocytoma
It is still unknown what causes the development of the disease. It was found that the tumor begins its development from chromaffin cells of the adrenal medulla. It is these cells that produce epinephrine (adrenaline) and norepinephrine (norepinephrine).
The risk of getting sick increases if the patient already has:
- multiple endocrine neoplasia type II or MEN II. Men suffer from this rare hereditary disease; tumors develop in the thyroid and parathyroid glands, on the lips, in the tongue, and in the gastrointestinal tract.
- Hippel-Landau disease, when multiple tumors occur in the nervous and endocrine systems, pancreas and kidneys.
- neurofibromatosis type 1: multiple tumors appear in the skin - neurofibromas, pigmented spots appear on the skin, and a tumor of the optic nerve also occurs.
- hereditary paraganglioma/pheochromocytoma syndrome, which occurs due to mutations in genes.
- Carney's triad: paragangliomas, pulmonary chondroma, gastric stromal tumor.
Why does the disease occur?
The pathogenesis of the disease in question is still not fully understood. The reasons contributing to the development of the pathological process in the adrenal glands include:
- Heredity. About ten percent have a family member with adrenal cancer. Geneticists believe that the disease pheochromocytoma is caused by disturbances in the functioning of chromosomes responsible for the production of hormones by the adrenal glands, which leads to uncontrolled growth of cells in the patient’s medulla;
- Formations of the endocrine nature of Gorlin syndrome (type 2B) and Sipple syndrome (type 2A), which are characterized by the proliferation of endocrine cells. In this case, the patient experiences damage to the musculoskeletal system, mucous membranes and thyroid gland.
Classification
The formation of the adrenal glands is distinguished, i.e. pheochromocytoma itself. In addition to the adrenal glands, a small number of chromaffin cells in the form of small clusters can be found in the heart, head, neck, bladder, and along the spine. Tumors in these cells are called paragangliomas. They have a similar effect on the body as pheochromocytomas, as the same hormones are released.
Classification of pheochromocytoma according to ICD-10
The disease is placed in section D35.0 - “Benign neoplasm of other and unspecified endocrine glands.”
Forecast
After removal of a benign tumor, the prognosis is favorable. After the operation, normalization of blood pressure and gradual regression of pathological symptoms are noted. The five-year survival rate in this case is 95%. In 10 cases out of 100, relapses are possible. In the postoperative period, you need to be under the supervision of an endocrinologist and undergo regular examinations (at least once a year).
In the absence of treatment, as well as after removal of a malignant tumor, the prognosis is less favorable. In this case, the patient can live no more than 5 years. When FCC is detected in pregnant women, the mortality rate is almost 50% (for both mother and child).
Symptoms and signs of adrenal pheochromocytoma
The main signs that will help suspect the disease are:
- elevated blood pressure levels
- increased, rapid, or irregular heartbeat
- excessive sweating for no reason
- severe headache
- pale face
- shortness of breath.
More rare symptoms may include:
- anxiety, depressing expectation of trouble
- abdominal pain, constipation
- weight loss
Catecholamine crises
The disease is characterized by the sudden onset of symptoms. These manifestations are called catecholamine crises. Their duration ranges from 15 to 20 minutes. The attack can occur several times a day or less often; in severe cases, there can be up to 25 crises per day. The attack usually ends as suddenly as it begins. During the period between crises, blood pressure may drop to normal or remain high.
Possible triggering factors for crises:
- physical exercise
- stress, nervous tension
- change in body position
- intestinal peristalsis
- contractions or childbirth.
Eating foods rich in tyramine can also trigger a crisis. This biogenic amine is abundant in fermented foods, for example:
- in mature cheeses
- in dried and smoked meat
- in avocados, bananas, beans
- in marinated fish
- in sauerkraut
- in some beers.
The release of hormones can be triggered by drugs (amphetamine, cocaine) and some medications:
- vasoconstrictors (decongestants)
- monoamine oxidase inhibitors
- dopamine receptor blockers
- serotonin reuptake inhibitors
- muscle relaxants
- glucocorticosteroids.
Clinical picture
The main symptom is arterial hypertension (constant, paroxysmal or mixed form). A characteristic feature of the hemodynamic (hypertensive) crisis in pheochromocytoma is its short duration and so-called self-limitation.
Additional symptoms include:
- orthostatic hypotension;
- sweating;
- constant headaches;
- feeling of inner trembling, anxiety;
- general weakness, decreased ability to work.
The classic course of pheochromocytoma is indicated when patients have hypertensive crises with a sudden increase, primarily in systolic blood pressure, which can reach 300 mm Hg. Crises can be provoked by slight physical activity, palpation of the abdomen, sometimes by taking β-blockers, and when pheochromocytoma is localized in the wall of the bladder, by urination; are accompanied by rapid heart rate (up to 180/min), arrhythmias and/or changes in the electrocardiogram (ECG) like acute coronary ischemia (usually not associated with coronary circulation disorders, but due to the direct toxic effect of catecholamines).
Also during an attack, tremors, tinnitus, anxiety or fear, dilated pupils, sweating, chest or abdominal pain, nausea or vomiting are often noted. Vasoconstriction of the extremities under the influence of catecholamines can cause pain and paresthesia, intermittent claudication, Raynaud's syndrome, ischemia, and trophic ulcers. Hyperglycemia and glycosuria, leukocytosis may be detected. The duration of an attack can be from several minutes (usually) to several hours (much less often). The attack usually ends suddenly. With pheochromocytoma, decompensation of previously undetected diabetes mellitus or impaired glucose tolerance is possible. When examining the fundus, spastic angiopathy is revealed. In pheochromocytoma, the severity of changes in the fundus does not correspond to the malignancy of the course of hypertension.
In approximately 10% of cases, pheochromocytoma is a familial disease and is inherited in an autosomal dominant manner. Pheochromocytomas and accompanying tumors of the thyroid gland and nervous tissue are of neuroectodermal origin, as evidenced by the presence of neuron-specific enolase in all these tumors. Apparently, the occurrence of such tumors is due to disturbances in the proliferation and differentiation of neural crest cells.
MEN type IIa. This hereditary syndrome is caused by a defect in one of the loci of chromosome 10. Components of the syndrome: medullary thyroid cancer, hyperplasia or adenoma of the parathyroid glands (clinically manifested as hyperparathyroidism), pheochromocytoma and (less commonly) bilateral adrenal hyperplasia.
MEN type IIb. EC components of the syndrome: pheochromocytoma, medullary thyroid cancer, mucosal neuromas, thickening of the corneal nerves, gastrointestinal ganglioneuromas; often Marfan-like appearance.
Other associated syndromes. 5% of patients with pheochromocytoma have neurofibromatosis (Recklinghausen's disease). A combination of pheochromocytoma, neurofibromatosis and somatostatin-containing carcinoid tumor of the duodenum has been described. A combination of pheochromocytoma with Hippel-Lindau disease (retinocerebellar hemangioblastomatosis) and acromegaly was observed.
Diagnosis of pheochromocytoma
The endocrinologist begins the diagnosis with questioning, finding out hereditary predisposition, and studying the history of the disease. Then he measures blood pressure and calculates heart rate.
Analyzes
Next, laboratory tests are usually prescribed to study the levels of adrenaline, norepinephrine and their metabolites:
- determination of metanephrine and normetanephrine in urine collected per day
- determination of the level of free metanephrine in blood plasma.
Measuring urinary catecholamine levels and vanillylmandelic acid levels is not currently used because false-positive results are common.
At elevated levels of biologically active substances, tests use imaging methods that will help identify the tumor:
- computed tomography (CT) of the abdominal organs and adrenal glands. The method allows you to detect formations larger than 5 mm.
- scintigraphy, which is used if there is a suspicion of a tumor outside the adrenal glands. When conducting research, it is necessary to take into account the possibility of asymmetric accumulation of the isotope in normal adrenal glands.
- positron emission tomography (PET). This method is more sensitive than scintigraphy in detecting metastatic lesions.
If several family members have the disease or developed before the age of 40, genetic testing is performed. For patients over 50 years of age, genetic testing is rarely prescribed.
Treatment of pheochromocytoma
In rare cases, taking medications can help control blood pressure. The main method of treatment is removal of the tumor.
Surgical treatment of pheochromocytoma
Adrenalectomy surgery is performed either laparoscopically, with the insertion of miniature surgical instruments through small incisions in the abdomen, or through open access to the tumor, when an incision is made in the skin of the abdomen. If the tumor is large (more than 8 cm), the open method is preferable, since during removal it is important not to damage the tumor capsule. Both types of operations are performed under general anesthesia.
If malignancy of pheochromocytoma is detected, the treatment options are as follows:
- surgical removal of the tumor
- radiation therapy
- chemotherapy
- tumor ablation using radio wave method or cryotherapy
- embolization therapy by blocking the artery supplying the tumor
- targeted therapy by blocking specific enzymes, proteins or molecules that affect tumor growth.
Medicines prescribed before surgery
To prepare for surgery, 7-14 days before the operation, the following is prescribed:
- alpha-blockers. These drugs reduce the effect of excess norepinephrine on the body and stabilize blood pressure by relaxing the vascular muscles. Doxazosin, prazosin, and phenoxybenzamine are usually used.
- beta blockers, which also lower blood pressure and heart rate by blocking the action of adrenaline. For example, atenolol, metoprolol and propranolol are used.
- calcium channel blockers, for example, diltiazem, amlodipine, nifidepine. Since diltiazem gently lowers blood pressure and has a relatively short duration of action, it is used for minor increases in blood pressure.
Often calcium channel blockers are added to alpha and beta blockers.
Emergency care for a sharp increase in blood pressure
If a patient with pheochromocytoma has a sharp increase in blood pressure, then an urgent need to call a doctor and, under his supervision, reduce the pressure. The following are used as emergency medications:
- phentolamine 5 to 20 mg intravenously
- tropafen 1%, 1-2 ml intravenously, very slowly
- sodium nitroprusside starting at 0.25 mcg per kilogram of body weight per minute IV.
- labetalol 100 mg orally. It is possible to administer intravenously at a dose of 20 mg, i.e. A 1% solution is administered in an amount of 2 ml.
The use of methyldopa, minoxidil, pentamine, and benzohexonium is undesirable.
Proper nutrition
The diet for pheochromocytoma should not stimulate the nervous system or speed up metabolism.
Proteins are strong catalysts for metabolism, so their quantity is limited, especially in the form of meat and fish. You can make up for lost protein with dairy products and eggs. Nutritionists recommend supplementing your diet with iodine by introducing seaweed, shrimp, and squid into your diet. Among vegetables, preference is given to cabbage, turnips, and zucchini. While on such a diet, you can carry out fasting days after 1-3 days. Salt is limited to 1-2 g per day. If you are overweight, you should reduce the amount of fat consumed and the total calorie content of food.
Complications of pheochromocytoma
Constant production of hormones increases blood pressure and can damage the heart, kidneys, and brain. Life-threatening conditions may occur, such as:
- arrhythmia
- myocardial infarction
- stroke
- renal failure
- acute respiratory distress syndrome.
Catecholamine shock
This complication is due to the fact that catecholamines are not inactivated and continue to act on the blood vessels. In this case, the change in pressure becomes unpredictable, episodes of increased pressure are chaotically replaced by hypotension. Alpha-blockers are used for treatment; in severe cases, hospitalization in the intensive care unit is necessary.
Malignant pheochromocytoma
Rarely, malignancy may occur, i.e. malignancy of education. This condition is called malignant pheochromocytoma. The diagnosis is confirmed by a biopsy, detection of metastases in the bones or other organs.