In medicine, Prader-Willi syndrome is a rare hereditary disease characterized by the absence or insufficient functioning of certain genes or their parts of the 15th paternal chromosome. This pathology was first described in 1956 in Switzerland by pediatricians A. Prader and H. Willi, after whose names the syndrome was named. Its frequency is 1 case per 12-15,000 newborns. Symptoms and manifestations of Prader-Willi syndrome vary, and the course of the disease usually depends on the specific case.
General information
Prader-Willi syndrome was first mentioned by Langdon Down in 1887 when describing a 14-year-old patient suffering from growth retardation, obesity, decreased mental activity and ovarian function (hypogonadism). Langdom Down called this disease polysartria.
The classic description of the syndrome appeared in 1956 thanks to studies of patients with a similar phenotype conducted by Swiss pediatricians Andrea Prader, Guido Fanconi, Heinrich Willi and physician Alexis Labhart.
In 1965, Angelman syndrome was identified, in which mutations in the same genetic region (15q11-13) affect not the paternal, but the maternal copy of the chromosome.
In 1981, while studying chromosome 15, Ledbetter and co-authors identified a region of the chromosome that, when mutated, causes Prader-Willi syndrome.
The incidence of the disease ranges from 1 in 10,000 to 1 in 25,000 newborns.
The disease is equally common in boys and girls, and is the main genetically determined cause of obesity in individuals over 1 year of age. Nationality and race do not affect the prevalence of the disease.
Prevention
It is impossible to prevent a congenital disease; the main thing in this case is to prevent complications from occurring. Treatment of the syndrome should begin as early as possible, then it will be easier for the child to adapt to school and life in society.
Prevention of the disease includes medical and genetic consultations for families who have a predisposition to the occurrence of the syndrome. Expectant parents need to conduct a prenatal genetic study, which will help determine the structural features of the fetal chromosomes.
To improve the life of a child with PWS, constant cooperation between medical specialists, parents and the child himself should be ensured.
Forms
In 2000, based on pathogenesis, researchers (Olaetser et al.) identified three phenotypes of Prader–Willi syndrome:
- a typical phenotype of Prader-Willi syndrome, which develops due to deletion of the paternal copy of the chromosome;
- a milder phenotype, characterized by better developed cognitive function (develops with uniparental maternal disomy);
- a pronounced phenotype with a large number of cardiac pathologies, developing with maternal uniparental disomy and mosaic 15 trisomy.
Notes
- Buiting K. et al.
Clinical utility gene card for: Prader-Willi Syndrome (English) // European Journal of Human Genetics. — 2014. — P. e1–e3. — DOI:10.1038/ejhg.2014.66. - Prader A., Labhart A., Willi H.
Ein Syndrome von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter (German) // Schweiz. Med. Wschr. - 1956. - Bd. 1986. - S. 1260-1261. - Swaab, D. F. et al. (1995) Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J. Clin. Endocrinol. Metab. 80, 573—579
Reasons for development
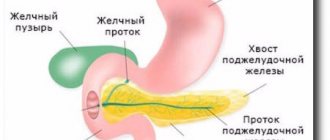
Prader-Willi syndrome occurs as a result of dysfunction of the q11-13 region of the 15th pair of chromosomes (PWS-AS region, associated with Prader-Willi and Angelman syndromes).
The development of Prader-Willi syndrome is always associated with a chromosome of paternal origin.
The disease develops when:
- Deletions (loss) of the q11-13 region of the paternal gamete (detected in 70% of cases).
- The complete absence of the paternal copy of the 15th chromosome and its replacement with a duplicated maternal copy (maternal disomy). Occurs in 20% of patients.
- Functional deactivation in the fetus as a result of methylation (modification of the molecule without changing the nucleotide sequence of DNA) of the structurally normal region q11-13 of the paternal chromosome. Occurs in 5% of patients.
The authors of the first description of the syndrome, based on observations, assumed that the disease has an autosomal recessive mode of inheritance. Subsequently observed familial cases of pathology gave reason to assume an autosomal dominant mode of inheritance, but most of the described cases were sporadic.
Further research revealed that Prader-Willi syndrome in children is associated either with translocations (chromosomal rearrangements in which different chromosomes exchange their fragments) or with mosaicism (the presence in tissues of cells that differ genetically).
The presence of microdeletion of chromosome 15 in Prader–Willi syndrome was established in 1987, and molecular genetic research methods made it possible to identify the role of genomic imprinting and uniparental disomy in the development of the disease.
For a long time it was believed that the expression (conversion of hereditary information into protein or RNA) of maternal and paternal genes is equivalent, but the identified phenomenon of genomic imprinting demonstrated the presence of selective expression of some chromosomal loci depending on their paternal or maternal origin.
The exact cause of uniparental disomy has not yet been established, but it has been discovered that the inheritance of two chromosomes from only one parent is the result of a series of genetic and biochemical disorders (for a long time such inheritance was considered impossible, but with the help of molecular genetic markers the possibility of such inheritance was established).
Diagnostic measures
Early diagnosis of Prader-Willi syndrome and subsequent treatment can improve the prognosis of the disease. The diagnosis is made, as a rule, on the basis of the clinical manifestations of the disease, but today genetic testing is often used, which experts recommend primarily for newborns. This is due to the fact that in children the presence of the syndrome is much more difficult to determine, since it is impossible to test their ability to diagnose Prader-Willi syndrome based on clinical manifestations. Genetic testing is carried out using the DNA methylation method to determine whether abnormalities are present on chromosome 15 that lead to the onset of the disease. This method of diagnosing Prader-Willi syndrome helps to identify 97% of cases of the disease. It is also worth noting that the disease is often misdiagnosed, since it is often confused with Down syndrome, which is much more common. In addition, such a characteristic sign of Prader-Willi syndrome as obesity may also be present in Down syndrome. For this reason, a huge number of cases of the disease remain undetected.
Pathogenesis
The pathogenesis of the disease remains poorly understood to date.
There are suggestions that obesity accompanying Prader-Willi syndrome is caused by very low processes of fat breakdown (lipolysis) and increased synthesis of fat from acetate by more than 10 times.
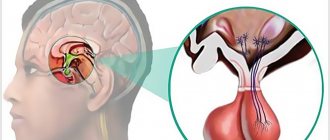
Hypogonadism, developing according to the hypogonadotropic type, is associated with dysfunction of the hypothalamus (presumably, the pathology affects the areas of the ventromedial and ventrolateral nuclei). This theory is confirmed by the effectiveness of treating patients with clomiphene, which promotes the secretion of gonadotropins.
The hypopigmentation of the skin, hair and iris characteristic of patients is explained by a decrease in the activity of tyrosinase, an enzyme containing copper and catalyzing the oxidation of phenols, in the hair follicles and in the cells that produce melanin.
The 2nd phase of the disease is characterized by a deficiency of growth hormone (GH) caused by dysfunction of the hypothalamus.
Prader–Willi syndrome occurs due to imprinting. The determination of the expression of alleles that occurs due to this process is associated with their origin (either the father's gene or the mother's gene is always expressed). As a result of the lack of expression of the alternative form of the gene (paternal), the pathogenic mutant allele of the mother is expressed in the child’s phenotype.
The beginning of the imprinting process is associated with the process of gamete formation. In the potentially inactive allele of the embryo genome, cytosine methylation occurs and CpG islands are formed in the DNA, which consist of guanine (G) and methylated cytosine (C) pairs. Due to methylation in the promoter region of the repressed gene, abortive (short, resulting from premature cessation of synthesis) RNA is formed, which is necessary for gene suppression. Thus, through methylation, the gene is marked for imprinting.
The occurrence of uniparental disomy is influenced by the age of the mother (it affects conceptions with trisomy or monosomy of chromosome 15). Such cases are explained by maternal nondisjunction, in which the homologous chromosomes of a pair do not separate, but are directed to one pole of the cell. After fertilization of such a gamete, a zygote is formed containing an odd number of chromosomes.
In many cases, the extra chromosome is removed before the embryo begins to develop. Since the mechanism for removing an extra chromosome does not take into account its origin, only in 66% of cases the cell gets rid of one of the maternal chromosomes, and in the rest the only paternal chromosome is removed.
Prader-Willi syndrome in 10% of cases occurs as a result of chromosomal rearrangement (translocation) in the region critical for the syndrome. Observations of families of patients with similar translocations showed a high risk of relapse (about 50%) and the dependence of the phenotype on the sex of the parent transmitting the chromosome.
The Prader–Willi syndrome phenotype occurs in 2–5% of cases due to imprinting defects. Defects that include division into two parts of the imprinting center within the 15th chromosome (region q11-q13) lead to a change in the epigenotype of the chromosome of one of the parents. These defects are often associated with deletion (loss) of the imprinting center, and in some cases with an epigenetic mutation that does not cause a change in the DNA nucleotide sequence.
In all cases, the identified defects lead to changes in DNA methylation, disturbances in chromatin structure and the content of transcripts of a given gene in various cells.
Deletions in the imprinting center are associated with a 50% risk of relapse (the risk depends on the sex of the parent who transmitted the anomaly), and in the absence of a deletion the risk of relapse is low.
Currently, genes that are affected by genomic imprinting on chromosome 15 (region q11-q13) have been identified only for Angelman syndrome. According to scientists, Prader–Willi syndrome is associated with genes in the cluster of non-coding small nucleolar RNAs (snoRNAs) or with protein-coding genes SNURF – SNRPN (located in the area of the imprinting center) and Necdin.
Endocrine disorders
There are several factors that support the concept of growth deficiency in individuals susceptible to the syndrome.
- Affected individuals are short and overly obese, meaning they have low free fat mass, low bone density, and low energy use.
- This disease is characterized by disturbances in the genitourinary system. In males, problems arise with undescended testicles (over time, the testicles may drop to normal levels), and in females, problems arise with the appearance of adrenarche. In both cases, it is possible to solve the problem surgically.
Symptoms
In the first year of life, identification of Prader-Willi syndrome is difficult as a result of the absence of specific symptoms at birth and the spontaneous elimination of early signs of the disease during the first months of life.
Children with Prader-Willi syndrome often experience slight intrauterine hypotrophy and asphyxia, and possible hip dysplasia. Babies are usually born full term. In 10-40% of children, breech presentation is observed.
Early manifestations of the syndrome include:
- weak fetal movements;
- decreased sucking reflex after birth (in some cases its absence is observed);
- muscle hypotonia developing in the first months of life.
If the act of sucking is disrupted, the absence of tube feeding can in a short time lead to the development of dystrophy and other deficiency conditions in the child.
Muscle hypotonia leads to impaired motor development.
During the first months of life, there is a spontaneous increase in muscle tone and restoration of the sucking reflex.
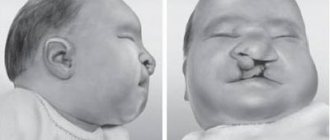
Already in the first year of life, various dysmorphia of the face and limbs may appear (acromicria, which is characterized by the presence of disproportionately small feet and hands).
Dysmorphia may be manifested by the presence of:
- elongated head shape or microcephaly;
- almond-shaped eyes;
- wide bridge of the nose;
- strabismus (strabismus);
- ectropion (inversion of the eyelid);
- thin upper lip and small mouth;
- low-lying ears and hypoplasia of ear cartilage.
Hypogonadism is also observed (boys are characterized by cryptorchidism, hypoplasia of the scrotum and penis, and girls have underdevelopment of the labia and in half the cases the uterus).
Children with Prader-Willi syndrome are characterized by increased sleepiness, the presence of thick saliva and dental problems (microdontia, etc.). In 75% of the total cases of the disease, weak pigmentation of the hair, iris and skin is observed.
Scoliosis and low bone density are also often detected.
Prader–Willi syndrome is usually diagnosed during the development of the second phase of the disease, which is characterized by:
- The appearance of a feeling of constant hunger and the formation of the behavior of “constant search for food” (polyphagia).
- Rapid development of obesity (fat deposits are observed mainly on the body and in the proximal limbs).
- Slowing down of growth processes. Without treatment, the final height of boys is 155 cm, and girls - 147 cm.
- The manifestation of moderate signs of intellectual impairment (with a normal IQ of 85-115 units, patients have from 20 to 80 units). Vocabulary is below normal, and patients' speech is usually difficult.
- Violation of the timing of sexual development and change in the order of appearance of secondary sexual characteristics.
The mood of patients with Prader-Willi symptom is characterized by frequent changes, but in general they are friendly.
In some cases, convulsions and loss of coordination occur.
The daytime sleepiness present in many cases occurs in some patients as narcolepsy, and episodes of night apnea are observed.
Diabetes mellitus may develop, with a tendency to improve with age.
NMR imaging in 12% of cases reveals abnormalities of the cerebral cortex and cysts of the cerebellar vermis.
Symptoms
The symptoms of Prader-Willi syndrome are different and vary depending on the age category of the patient.
In utero the pathological condition has the following symptoms:
- decreased activity of fetal movements;
- unusual location;
- polyhydramnios.
Immediately after birth, symptoms are as follows:
- breech presentation of the fetus;
- low pressure;
- decreased sucking reflex;
- labored breathing.
During childhood, the child experiences the following conditions:
- inhibited development of speech skills;
- damage to teeth by caries;
- depressed coordination of movements;
- eating large amounts of food;
- rapid weight gain;
- sleep problems;
- scoliosis;
- puberty occurs later than usual;
- short stature;
- delayed intellectual development;
- delayed psychomotor development;
- excessive flexibility.
Signs in adulthood:
- infertility;
- a small amount of hair in the pubic area;
- obesity;
- tendency to diabetes.
Generalized symptoms of external differences in adults include:
- large, wide nose;
- narrow fingers;
- small limbs;
- there is hypersensitivity of the skin;
- a large amount of excess weight;
- forehead high and narrow;
- skin and hair are lighter in color compared to relatives.
There is a delay in motor and sexual development.
Diagnostics
Since identifying the Prader-Willi symptom before the 2nd phase of the disease allows the child to develop correct eating behavior, and correction of growth hormone deficiency begun before 18 months of age contributes to the formation of a correct physique, early diagnosis of the symptom is very important.
The diagnosis is based on an assessment of the patient’s symptoms, which are divided into major and minor. Prader-Willi syndrome is assumed in children under 3 years of age if there are at least 5 points, and after 3 years - if there are 8 points, of which 4 are considered major signs of the disease.
Major features worth 1 point include:
- Characteristic facial features. These features include dolichocephaly with a decrease in diameter in the area of the temporal bones, almond-shaped eyes and strabismus, a small mouth with downward corners, and a thin upper lip.
- Delayed neuropsychic development under the age of 6 years and moderate decline in intelligence.
- Problems with feeding in the first months of life, followed by normalization of sucking.
- Changes in the sexual sphere.
- Muscular hypotonia of central origin, which was present in early childhood.
- Obesity progressing from 1 year to 6 years.
Minor signs (0.5 points each) include:
- reduced motor activity of the fetus and the presence of infantile lethargy;
- refractive errors;
- decreased skin and hair pigmentation compared to parents;
- “attrition” of the skin;
- the presence of a transverse palmar fold;
- short stature at age 15 compared to other family members;
- the presence of sleep disorders and episodes of apnea;
- small size of feet and hands;
- presence of articulation and speech defects;
- saliva viscosity;
- behavioral disorders.
Despite the high sensitivity of the scale, diagnosis requires karyotyping (in most cases the karyotype is normal) and molecular genetic studies of the 15th pair of chromosomes.
To identify microdeletion or uniparental disomy, methods of prometaphase analysis, the use of DNA markers of certain sections of chromosome 15, etc. are used.
A differential diagnosis is required with Lawrence-Moon-Bardet-Biedl syndrome, adiposogenital dystrophy, congenital myopathy, congenital spinal amyotrophy and Alström syndrome.
Material and methods
All patients were sent to the laboratories of the Scientific Center for Mental Health of the Russian Academy of Medical Sciences and the Moscow Research Institute of Pediatrics and Pediatric Surgery to confirm the diagnosis.
Preliminary genetic analysis of sick children was carried out using various cytogenetic and molecular cytogenetic diagnostic tests for the most common gene and chromosomal syndromes associated with autistic disorders and syndromic forms of mental retardation, including FISH to analyze cases of chromosomal mosaicism and subtelomeric microdeletions and duplications [1, 4, 13, 15-22]. Molecular karyotyping in this cohort of children was described by us previously [5, 6, 10]. In this work, cytogenomic studies were carried out on 30 children with a presumptive diagnosis of Angelman or Prader-Willi syndromes.
In silico
or bioinformatics analysis of identified genome variations (CNVs) was carried out using the following databases: DECIPHER (database of unbalanced chromosome aberrations) - decipher.sanger.ac.uk, OMIM (online Mendelian inheritance in Man) - www.omim.org, The Phenotype -Genotype Integrator (PheGenI) - www.ncbi.nlm.nih.gov/gap/PheGenI, SFARI Gene/AutDB (web-based searchable database for autism research) - www.mindspec.org/autdb.html, Catalog of Published Genome -Wide Association Studies (NHGRI) www.genome.gov/gwastudies.
Treatment
Since Prader-Willi syndrome is a congenital genetic abnormality, there are no specific treatments for it.
Therapy is aimed at normalizing the metabolism of fats and carbohydrates, as well as eliminating the symptoms of the disease.
When treating patients, the following are used:
- diet therapy, including recording the calorie content of food and monitoring the diet;
- oral hypoglycemic drugs for the treatment of diabetes mellitus;
- clomiphene, which restores the secretion of gonadotropins and sex steroids;
- a growth hormone.
At an early stage of the disease, massage is prescribed for hypotonicity.
If disorders of bioenergy metabolism are detected, vitamins (E, C, B2, thiamine, vitamin PP), coenzyme Q10, and cinnarizine, which improves blood circulation, are prescribed.
Drug treatment is supplemented by classes with a speech therapist and speech pathologist.
Children with Prader-Willi syndrome are constantly under the supervision of an endocrinologist, neurologist, psychotherapist and ophthalmologist.
Life expectancy forecast
The severity of the disease depends on how much it is possible to compensate for disturbances in carbohydrate and fat metabolism with diet and medications. Diabetes mellitus and obesity lead to severe vascular complications, heart attack, and stroke. They are associated with major life risks.
The ability of DNA to self-repair is reduced; in old age, this leads to an increased risk of tumor processes. With good health control, patients live to 65 years or more .