Acute thrombocytopenic purpura
Acute thrombocytopenic purpura (syn. post-infectious, or haptenic, Thrombocytopenic purpura
).
Etiology and pathogenesis
Thrombocytopenia (see) develops due to increased destruction of blood platelets. The reasons for the increased destruction of platelets (see) in this form of P. t. have not been studied enough. The main importance is attached to the excess in the blood of antigen-antibody complexes (see Antigen-antibody reaction), formed in response to the introduction of the virus. The binding of immune complexes to a certain locus of the platelet membrane (Fc receptor) creates the preconditions for the death of the latter. Platelets loaded with immune complexes are phagocytosed in the spleen and liver or destroyed under the influence of complement (see) directly in the bloodstream. It is likely that platelets can be destroyed by antibodies directed against virus-modified platelet autoantigens. After the virus and its antigens are removed from the body, antibody formation stops, the level of platelet destruction normalizes, and recovery occurs.
Clinical picture
It is observed mainly in children of preschool and primary school age. It starts off sharp.
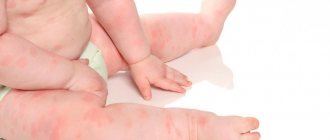
The clinical manifestation of the disease in 80-85% of cases is preceded by a period of fever, often associated with viral infections such as rubella (see), measles (see), chicken pox (see), influenza (see). Cases of P. t. after vaccination are described. The interval from the onset of infection to the onset of P. t. ranges from 3 days to 3 weeks. Characterized by rashes of various sizes, including petechial ones, and bleeding gums. Stomach and kidney bleeding are possible. In 10-20% of cases there is a slight enlargement of the spleen and liver. 60% of children have moderate lymphadenopathy. Brain hemorrhages are possible in the first 2 weeks. diseases, especially in the presence of petechiae on the face, hemorrhages in the oral mucosa and conjunctiva. Deep thrombocytopenia is usually recorded in the blood (less than 20,000 in 1 μl), in 80% of cases - relative lymphocytosis, in 20% - eosinophilia. Radioisotope studies show a sharp shortening in the lifespan of platelets, sometimes up to several hours. A compensatory increase in their production is accompanied by an increase in the number of megakaryocytes in the bone marrow and the appearance of giant forms of blood platelets in the blood.
Diagnosis
The diagnosis is based on the exclusion of symptomatic forms of P. t. and usually does not cause difficulties. The immune nature of the disease is confirmed by the detection of increased levels of immunoglobulin class G (antibodies) on the surface of platelets compared to the norm.
Treatment and prognosis
Prednisolone is prescribed at a dose of 1-3 mg/kg body weight (weight) for 3-4 weeks. According to Lasher and Iyer (J. M. Lusher, R. Iyer), glucocorticoid hormones do not reduce the duration of the disease and usually do not change the level of platelets, but they relieve hemorrhagic syndrome and reduce the likelihood of cerebral hemorrhage. Platelet transfusions are most often ineffective; transfusion of large quantities of platelets sometimes stops bleeding.
The disease lasts several weeks or months; usually ends with spontaneous recovery after 1-2 months. (maximum 6 months); recovery is observed in 80% of patients; in the rest it becomes chronic. Mortality approx. 1%. It is advisable to refrain from splenectomy during this period, except in cases where the operation should be performed for vital indications (crisis that cannot be controlled by large doses of prednisolone, severe hemorrhagic syndrome, the appearance of symptoms indicating the possibility of hemorrhage in the brain). Cytostatic therapy for this form of P. t. is inappropriate. If thrombocytopenia persists for more than 6 months, one can think of a chronic form of the disease (see below).
Prevention of bleeding
in the acute form of P., t. consists of limiting mobility at the height of the disease, excluding the possibility of injury. Taking medications that interfere with platelet function is contraindicated.
General information
Thrombocytopenic purpura is a group of hematological diseases of various etiologies, which boils down to a decrease in the number of blood platelets - platelets below 150x10 to the ninth power per liter of blood, caused by a violation of the platelet component of hemostasis . Most often it develops due to autoimmune processes of exposure to antiplatelet antibodies and destruction by macrophages .
Chronic thrombocytopenic purpura
Chronic thrombocytopenic purpura (syn. autoimmune P. t.). This form of P. t. was first described in 1735 by PG Werlhof. Frank (A. E. Frank, 1915) was the first to suggest that with P. t. platelet production decreases under the influence of a factor produced by the spleen. For almost half a century, this hypothesis remained popular, but not confirmed. An alternative version belonged to Katznelson (P. Katznelson, 1916), who noted the positive effect of splenectomy in P. t. and on this basis argued that thrombocytopenia develops as a result of increased destruction of platelets in the spleen; this has been confirmed by numerous studies over the past two decades.
Etiology and pathogenesis
The causes of the disease are unknown. In the classical experiments of Harrington (WJ Harrington, 1951-1953), carried out by him and his laboratory staff, it was found that the gamma globulin fraction of the blood plasma in 60% of patients with P. t. contains a factor, the introduction of which to healthy people induces thrombocytopenia. Subsequently, it was shown that the antiplatelet factor is an immunoglobulin of class G (see Immunoglobulins), that it is able to interact with platelets as an antibody and can be detected on the surface of the patient’s blood plates in 95% of cases. McMillan (R. McMillan) and his colleagues proved that antiplatelet IgG, synthesized in vitro by lymphocytes of patients with P. t., is able to bind not only to its own, but also to donor, i.e., platelets that are not antigenically changed (see .). Both donor and autologous platelets have mitogenic activity and are capable of causing blast transformation of lymphocytes of patients with P. t. The listed data were the basis for classifying this form of P. t. to the group of the so-called. autoimmune diseases (see Autoallergic diseases). Pathogenesis of long-term persistent thrombocytopenia, in which immunol. samples turn out to be negative; it has not been definitively established. Antiplatelet (antibodies), by binding to platelets, not only disrupts the function of the latter, but also causes their subsequent destruction in the spleen and liver. The specificity of antiplatelet antibodies in autoimmune thrombocytopenia is unknown, but it has been proven that in different patients they are fixed to different loci of the platelet membrane. Increased platelet destruction is confirmed by a significant shortening of the lifespan of isotope-labeled cells. At steady state, the level of platelet production is 2-8 times higher than normal. This process is reflected in the increase in the number and (or) size of megakaryocytes in the bone marrow and the appearance in the blood of immature, enlarged platelets (see Bone Marrow). Increased bleeding in P. t. is associated both with a deficiency of blood platelets and with a disruption of their function under the influence of antibodies. That is why in this disease, unlike amegakaryocytic thrombocytopenia, in which the severity of the hemorrhagic syndrome clearly correlates with the degree of platelet deficiency, significant bleeding can be observed with a moderate decrease in platelet levels.
Pathological anatomy
The main object of pathological examination is the removed spleen (see). When performing a splenectomy (see) and suspicion of chronic active hepatitis (see) and the symptomatic nature of the deficiency of blood platelets, a marginal liver biopsy is performed (see Liver, research methods).
The spleen in most cases has normal size and weight, sometimes it is enlarged (up to 400 g). Microscopically, lymph tissue hyperplasia is often detected, with an increased number and size of follicles, they contain large germinal centers surrounded by a ring of mature lymphocytes, adjoined by a wide marginal zone of immature lymphoid elements. In some cases, the germinal centers are poor in cells, their tissue is impregnated with protein masses; Such changes are observed mainly with long-term use of corticosteroid hormones. In the red pulp of the spleen there is often an increase in the number of eosinophils and plasma cells; accumulations of nuclear forms of red cells and megakaryocytes can be detected; in germinal centers, marginal zones and pulp cords, as a rule, the number of mononuclear phagocytes is increased. Sometimes groups of xanthoma cells (lipoid macrophages, Gaucher-like cells) are found in the spleen tissue, which are apparently formed as a result of phagocytosis of destroyed platelets by macrophages. There is often a massive accumulation of protein substances in the walls of the central and pulp arteries, sinus membranes with their thickening and subsequent sclerosis. The endothelium of the sinuses is hyperplastic.
In the liver, protein and fatty degeneration of hepatocytes, expressed in varying degrees of hyperplasia of stellate endothelial cells, are detected. Trephine biopsy (see) of the iliac crest reveals bone marrow that is polymorphic in cellular composition, a significant increase in the number of megakaryocytes with the presence of immature forms among them. In cases of massive repeated bleeding or concomitant hemolysis, an increase in the nuclear elements of the red row cells may be observed.
Clinical picture
It is more often observed at 20-50 years of age, less often in children. Women get sick 3-4 times more often than men. The disease manifests itself gradually, gradually, less often acutely, and a connection with various provoking factors (viral or bacterial infection, excessive insolation, trauma) can be detected. Thrombocytopenic hemorrhagic syndrome is characterized by skin manifestations (ecchymosis, petechiae, extravasation at injection sites) and bleeding from the mucous membranes. The appearance of hemorrhages on the face, in the conjunctiva of the eyes, on the lips is a serious symptom indicating the possibility of hemorrhage in the brain (see Stroke). Bleeding from the gastrointestinal tract. tract, hematuria, hemoptysis are observed less frequently. An increase in the size of the spleen is not typical. With splenomegaly (see), even minor, it is necessary to exclude the symptomatic nature of the deficiency of blood platelets. In rare cases, the platelet count will normalize spontaneously. Usually this is observed no later than 6 months from the moment the first symptoms of the disease appear, i.e. we can talk about acute (post-infectious) P. t. (see above).
Thrombocytopenia of varying severity is recorded in the blood (usually the number of platelets does not exceed 75,000 in 1 μl); an increased content of giant forms of platelets with blue cytoplasm is often detected. When the platelet content is above 50,000 per 1 μl, hemorrhagic syndrome is rarely observed. Bone marrow examination reveals an increased or normal number of megakaryocytes. A significant part of them are represented by young forms with increased sizes and basophilic cytoplasm. The absence of platelets around megakaryocytes is not evidence of impaired release of platelet subunits, but indicates an accelerated entry of cells into the blood or their destruction in the bone marrow. Sometimes there is irritation of the red germ in the bone marrow associated with bleeding. Erythroid hyperplasia can be caused by concomitant hemolysis (see), which is confirmed by the detection of anti-erythrocyte antibodies using the Coombs antiglobulin test (see Coombs reaction). Bleeding time (see) is prolonged. Blood clot retraction is reduced (see Retraction). Blood clotting is normal in most cases (see Blood coagulation system). Sometimes fibrinogen degradation products are found in the blood. In some patients, a decrease in platelet adhesion to glass and a violation of adenosine diphosphate, thrombin, and collagen aggregation are detected. According to V. G. Savchenko and L. I. Idelson (1981), with immunol. The study determines an increased level of IgG on the surface of the patient’s platelets compared to the norm, which indirectly confirms the presence of antiplatelet antibodies.
Diagnosis
The diagnosis is based on the exclusion of hereditary and symptomatic forms of P. t. (see Thrombocytopenia). In diagnostically difficult situations, the method for quantitative determination of IgG on the surface of autologous platelets, developed by Dixon and Rosse (R. Dixon, W. Rosse), can be used. Indirect (serum) methods are currently practically not used due to low information content. It is impossible to differentiate acute (post-infectious) P. t. from chronic (autoimmune) using immunological methods in the early stages. When making a diagnosis, take into account that acute (post-infectious) thrombocytopenic purpura is observed mainly in children, lasts several weeks or months and usually ends with spontaneous recovery after 1 - 2, maximum 6 months.
Laboratory research
Characterized by a decrease in the content of platelets in the blood, up to single ones in the drug, and an increase in bleeding time. The duration of bleeding does not always correspond to the degree of thrombocytopenia, since it depends not only on the number of platelets, but also on their qualitative characteristics. Retraction of the blood clot is significantly reduced or does not occur at all. Secondarily (as a result of thrombocytopenia), the plasma-coagulation properties of the blood change, which is manifested by insufficient thromboplastin formation due to a deficiency of the 3rd platelet factor. Impaired thromboplastin formation leads to a decrease in prothrombin consumption during blood coagulation. In some cases, with thrombocytopenic purpura during a crisis, activation of the fibrinolytic system and an increase in anticoagulant activity (antithrombins, heparin) are noted. All patients with thrombocytopenia have a reduced concentration of serotonin in the blood. Endothelial tests (tourniquet, pinch, hammer, prick) during a hematological crisis are positive. No changes are found in the red blood and leukogram (in the absence of blood loss). Examination of red bone marrow usually reveals normal or increased levels of megakaryocytes.
Diagnostics
The examination is the task of a hematologist. As a rule, the process is primary, so it is almost never necessary to involve other doctors. With rare exceptions.
The doctor evaluates the clinical data. Patient complaints about health and well-being. This allows you to get an idea of the symptoms, put a set of signs under certain schemes, and put forward hypotheses.
It is also important to take an anamnesis. Previous and current diseases, characteristics of the mother’s pregnancy (if such moments are known), bad habits and other facts.
Both routine studies play an important role in primary diagnosis. Then the actual identification of the pathological process begins.
- General blood analysis. Gives an idea of the concentration of platelets in the biomaterial. A reliable and informative method, despite its simplicity.
- Coagulogram. The rate of coagulation of liquid tissue is subject to assessment.
- If necessary, an immunological test or ELISA is prescribed.
- In complex and controversial cases, a bone marrow puncture is performed. Usually the diagnosis is obvious even without such radical measures.
Diagnosis is relatively simple. The disease is detected and classified based on a combination of objective data and information received from the patient regarding medical history and complaints.
Etiology of the disease
In most cases, it is impossible to determine the causes of thrombocytopenic purpura, although scientists have been able to find out that genetic defects do not play a major role in its development. Sometimes it occurs against the background of hereditary pathologies associated with impaired production of thrombocytopoietins in the body or a decrease in enzymes included in the Krebs cycle, but these are isolated cases.
The most likely causes that can cause purpura, doctors include:
- disruptions in the functioning of the hematopoietic system caused by the development of aplastic anemia;
- tumor lesions of the bone marrow;
- the harmful effects of radiation on the human body, which causes disruption of myelopoiesis - the process of development and maturation of blood cells;
- a previous bacterial or viral infection (statistically, in 40% of cases Werlhof’s disease occurs precisely because of this);
- vascular replacement surgeries that could cause mechanical damage to platelets;
- pathological reaction of the body to the administration of gamma globulin;
- use of certain contraceptive drugs;
- chemotherapy for the treatment of cancer.
Sometimes the cause of the development of the disease in adults is collagenosis of an autoimmune nature, prolonged blood stagnation or pregnancy (this fact has been confirmed by few scientists and doctors, so it is rarely mentioned).
Treatment
Therapy is predominantly medicinal. Several types of medications are used.
- Corticosteroids. Prednisolone, in more complex cases - Dexamethasone, as a powerful analogue. To relieve inflammatory processes and inhibit the autoimmune reaction. Clinical manifestations with the use of such drugs are sharply weakened.
- Hyperimmune globulins. appointed for the same purpose.
- Cytostatics are used. Typically this category is indicated as part of the treatment of cancer. They slow down the growth of so-called “fast” cells. Hair, nails, tumors.
These also include the structures of the body’s defenses. Immune thrombocytopenic purpura is always treated with drugs of this group, without them there is no point in therapy, specific names are selected by the doctor. Such medications are extremely dangerous.
In case of severe dysfunction of the blood, transfusion of plasma or liquid fraction is possible. According to indications.
Conservative methods are not always effective enough. In such a situation, removal of the spleen becomes a promising option.
According to studies and statistical data obtained, the probability of a favorable outcome with such therapy reaches almost 70%.
Treatment is focused on the characteristics in each specific case, especially since there are several forms of purpura.
During pregnancy
Thrombocytopenic purpura usually poses a threat to both mother and fetus, especially if frequent relapses occur. Pregnancy causes its exacerbation more often in the second or third trimester. gestosis develops .
The risk of miscarriage is quite high - approximately 25%, placental abruption is possible. Most often, weak labor and postpartum hemorrhage are detected.
Complications in the fetus and newborn child are expressed in the form of deep prematurity, intracranial hemorrhages, malnutrition and hypoxia , anemia in newborns, postpartum petechial hemorrhage, melena , hematuria .
Normal platelet count in blood
| Age | Value, thousand/µl |
| Children | |
| Up to 1 year | 160-390 |
| 1 year – 10 years | 180-320 |
| 10 – 16 years old | 180-350 |
| Men | |
| Over 16 years old | 200-400 |
| Women | |
| up to 16 years old | 180-340 |
| Menstruation and the week after it ends | 75-220 |
| Healthy woman after 16 years | 180-320 |
| Pregnancy | 100-420 |
Risk factors
Thrombocytopenic purpura can occur in children and adults of any age, but there are factors that increase the risk of developing this condition:
- being female (thrombocytopenic purpura occurs three times more often in women than in men);
- a recent infectious disease increases the risk of developing the disease, especially in children;
- heredity (the disease in close relatives increases the risk of thrombocytopenia);
- frequent stress.
Consequences and complications
A chronic decrease in the level of platelets in the peripheral bloodstream can lead to serious conditions and diseases:
- massive blood loss ;
- hypochromic anemia – decrease in hemoglobin content less than 30 pkg;
- sideropenic state - a complex of clinical manifestations caused by iron deficiency with progressive anemia, characterized by peeling and dry skin, atrophy of the epithelium of the tongue, nose, pharynx and esophagus, the development of atrophic gastritis , muscle weakness, weakness of the urinary sphincter, gastrointestinal atony, impaired contractile function of the heart, fainting , decreased resistance to infections.
Causes
Part of the issue has already been raised earlier. If we talk about specific provoking factors:
- Burdened family history. The influence of this point has not been fully studied, but we can already talk about increased risks if one or, especially, several relatives in the ascending line had a history of thrombocytic purpura.
- Taking certain medications. Anti-inflammatory non-steroidal origin, antibiotics and others. There are many options. The dosage form has an unstable course and is prone to self-regression after some time.
- Infectious processes of any localization. Viral, bacterial or fungal. Strains of herpes and pyogenic flora (for example, Staphylococcus aureus) are especially dangerous.
- Surgical interventions performed. In the recent past. In this way, the body can respond to a traumatic factor from the outside.
- Blood transfusion. Relatively rarely, thrombocytopenic purpura of this origin causes critical disorders. This is usually a transient disorder, but its course is acute. and requires inpatient care.
- Direct transfer of antibodies to the fetus.
- Immunological incompatibility of mother and child.
- Autoimmune processes. Inflammatory. According to the type of lupus erythematosus, rheumatism, arthritis, thyroiditis (damage to the thyroid gland). The question is not in localization, but in the very essence of the phenomenon.
Such pathological processes are accompanied by hypersensitization of the body, increased sensitivity to the slightest influence.
Anything can trigger purple. From an allergen in food to a cold or prolonged exposure to the sun.
- Congenital bone marrow pathologies. Lead to insufficient platelet synthesis. This is a chronic disorder and requires constant drug treatment and regular examinations by a hematologist. Leukemia, malignant tumors.
The reasons are always pathogenic. But not in all cases it is possible to identify the culprit of the violation.
Doctors often talk about the idiopathic form, then therapy comes down to relieving symptoms and preventing progression.
Prevention
Since the mechanisms and causes of Werlhof's disease have not been sufficiently studied at present, primary prevention means that could completely prevent the development of the disease have not been developed. Secondary prevention, aimed at preventing relapses and exacerbations of the disease, consists of:
- minimizing the risk of injury;
- avoiding contact with infectious patients;
- diet (exclude spicy foods, alcohol, caffeine);
- avoiding direct sunlight, solarium, and some physiotherapy procedures (UHF, UV radiation).
- exemption from physical education.
Werlhof's disease is a serious disease that can lead to various complications. Therefore, parents need to closely monitor the well-being of their child. If any symptoms appear, you should consult a doctor as soon as possible. Following the recommendations of a specialist will help keep your baby healthy.