Hemorrhagic fevers
Pathogenesis of hemorrhagic conditions in hemorrhagic fevers. Thrombohemorrhagic syndrome (Machabeli syndrome) is an important link in the pathogenesis of many infectious diseases. Thrombohemorrhagic syndrome is a symptom complex caused by the universal and nonspecific property of blood, lymph, tissue fluid, cellular and intercellular structures to thicken reversibly and irreversibly due to the activation of their ability to coagulate and, as a result of retraction, to stratify into components of different states of aggregation. Thrombohemorrhagic syndrome goes through 4 stages in its development. The hypercoagulation stage begins in the tissue cells of the damaged organ, which leads to the release of coagulation-active substances; the coagulation activation reaction spreads to the blood. This stage is usually short-lived.
Stage of increasing consumption coagulopathy , inconsistent fibripolytic activity. It is characterized by a decrease in the number of platelets and fibrinogen levels, as well as the consumption of other plasma factors of the body’s coagulation-lytic system. This is the stage of incipient and increasing DIC (incomplete DIC syndrome).
The stage of defibrinogenation and total, but not constant fibrinolysis (defibrinogenation-fibrinolytic). A synonym for this stage is complete DIC syndrome.
The recovery stage or the stage of residual thrombosis and occlusion. With a favorable course of the syndrome, there is a return to physiological norms of all factors of the body's coagulation-lytic system.
According to clinical manifestations, various forms of thrombohemorrhagic syndrome are distinguished (fulminant, acute, chronic, latent, hemorrhagic, hyperergic, etc.). Thrombohemorrhagic syndrome is observed in trauma, surgical diseases, oncology, obstetric pathology, sepsis and many infectious diseases. In this chapter we consider only the features of the pathogenesis of hemorrhagic syndrome in hemorrhagic fevers.
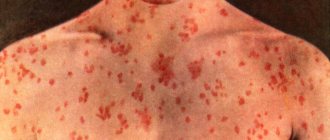
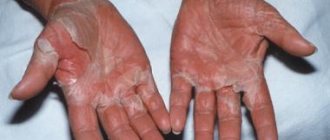
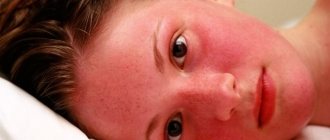
In hemorrhagic fevers, the primary pathology of suffering develops at the cellular-molecular level with the obligatory involvement of endothelial cells of the circulatory system and pluripotent stem cells of the bone marrow in the infectious process. The rate of development of the process is determined by the aggressiveness of the pathogen and its tropism towards other sensitive cells, for example, macrophages-monocytes, as well as the degree of maturity of the immune system of the individual affected by a specific pathogen. As a result, the normal physiological level of functioning of the endothelium and hematopoietic organs changes dynamically. At the height of the infection, a total change occurs in the anatomical and morphological integrity and, accordingly, the functions of the blood microcirculation system. It is accompanied by a deterioration in the trophism of all organs and tissues, hypoxia and their functional failure develop. The process is accompanied by disruption of the activity of the thermoregulatory and coordinating centers of the central nervous system, cardiovascular and excretory systems, pulmonary or other organ pathology develops, and patients often die in a coma. Life-threatening volumetric blood loss is rare, although increased bleeding, as a sign of changes in vascular permeability, acts as one of the leading symptoms of hemorrhagic viral fevers.
Among the causes of hemorrhagic syndrome, which develops quite quickly and often ends in the death of patients, viruses of five families are known: Arena-, Bunya-, Filo-, Flavi- and Togaviridae. They include the Lassa, Junin, Machupo, Guanarito, Sebia viruses (arenavirus family) - the causative agents of Lassa, Argentine, Bolivian, Venezuelan and Brazilian fevers, respectively; Rift Valley fever and Crimean Congo fever (bunyavirus family); yellow fever (flavivirus family); Marburg and Ebola fevers (filovirus family), dengue fever, Kyasanur forest disease and hemorrhagic fever with renal syndrome (togavirus family). The causative agents of tick-borne rickettsiosis, ehrlichiosis and the typhus group, on the contrary, are involved in hemorrhagic syndrome with a slower and mostly benign course. Consequently, in terms of attachment to the causative agent of an infectious disease, hemorrhagic syndrome is polyetiological.
A wide range of pathogens that cause a clinically uniform set of symptoms is due to the same type of mechanism of its formation. From the data of morphological and immunohistological studies of the above diseases, it is known that the main pathology unfolds in the endothelial cells of the microcirculatory system, bone marrow cells, and for some infections (Marburg fever, dengue), mononuclear phagocytes are involved in the process, both circulating in the bloodstream and tissue. This is reflected in the cellular, protein and peptide (kinins, leukotrienes) composition of the blood and the balance of its biologically active components. The activity of the complement, kinin, coagulation and anticoagulation systems is affected. The latter regulate vascular tone, their permeability, secretory activity of the endothelium, rheological properties of blood and are responsible for the subjective sensations of pain, fatigue and other symptoms of diseases.
Despite the etiological diversity of pathogens associated with the development of hemorrhagic syndrome, they are united by an identical or very similar mechanism for delivering the pathogen to target cells at the initial stage of the infectious process. The causative agents of fevers, transmitted with the participation of blood-sucking carriers, contact through scratching of the skin or aerogenously through the alveolar-capillary membranes of the lungs, are essentially mechanically transferred to pathogen-sensitive endothelial cells and tissue resident macrophages in places of microtrauma caused by the piercing-sucking apparatus of carriers or damage to the epithelium. It is at these points or in the alveolar-capillary membranes, on which particles of the infectious aerosol are retained, that the primary local process is formed. The protective reaction at the site of the entry gate is accompanied by the simultaneous generalization of the infectious process due to the dissemination of the pathogen with blood and lymph drainage with further consistently increasing damage to target cells in organs and tissues remote from the site of the entry gate of infection. In this case, some of the pathogen particles are absorbed by macrophages and other blood cells, for example, erythrocytes in rickettsiosis and bartonellosis, but are not inactivated due to insufficient concentration of cytokines, such as interferongamma (INF-y), tumor necrosis factor-alpha (TNF-a), and other monokines and the absence of specific neutralizing antibodies in the blood plasma at the time of infection and in the initial period of the disease.
Local damage to physiologically highly active cells that regulate hemostasis in the area of the entrance gate of the pathogen and the simultaneous generalization of infection with a transmissible mechanism of transmission are predetermined by the blood-sucking characteristics of ticks and mosquitoes, carriers of the pathogens of most hemorrhagic fevers, and by the peculiarities of the anatomical structure of tissue and alveolar capillaries. The synchronicity of the formation of local and generalized processes is explained by the fact that blood sucking in vectors lasts long and intermittently, especially in ticks (up to several days). Periodic suction of blood is accompanied by periodic injection into the capillary of saliva, and in some cases, coxal fluid containing the pathogen, anticoagulants and bloodsucker enzymes.
The infectious process at the level of the pathogen cell begins according to the usual scheme for intracellular parasites: attachment to the surface membrane and its loosening, penetration into the cytoplasm, reproduction or death (in the case of abortive infection), exit into the surrounding intercellular space or bloodstream, with infection of adjacent and distant intact cells. The last stage of cell-pathogen interaction is accompanied by either destruction and necrolysis of the first, or an increase in intracellular physiological processes with increased production of cytokines and other metabolites of their normal functioning.
The process of overcoming the cell wall by a pathogen is not indifferent to the macroorganism: membrane lipopolysaccharides are broken down by phospholipases of the carrier and pathogen. As a result, arachidonic acid is released - the substrate precursor of the biologically most active eicosanoids (prostaglandin E2 (PGE9), thrombooxanes, platelet activating factor (PAF), interleukin-I (IL-1) and others), responsible in the macroorganism for the tone and permeability of small arterioles and precapillaries, chemotaxis of leukocytes, thermoregulation, pain and other reactions of the body, united under the general name “endogenous toxicosis”.
Reproduction of the pathogen in endothelial cells, according to electron microscopic observations, is accompanied by their swelling and vacuolization, and then by obvious pathology - detachment from the basement membrane in the areas of capillary venules or from underlying cells in larger vessels and the appearance of some endothelial cells in the bloodstream. At the junction of the capillary venule, anatomical defects occur with exposure of part of the basement membrane. The weakness of the vascular walls in these loci with a progressive excess of PGE2, PAF and other cytokines causes a persistent expansion of the lumen of the venules with an increase in their blood supply; blood flow slows down, leukocyte-platelet stasis, peripheral hypotension, and then edema and hemorrhage form due to plasma exudation and migration of blood cells into the perivascular space from postcapillary venules. Initially, the exudation process is compensated, but the increase in the infectious process as a whole with the expansion of destructive-inflammatory changes in the vascular and macrophage systems overcomes the “threshold” of compensatory protective reactions of the body. The latter is obviously determined by the characteristics of the constitutional immunity of the patient, the age-related degree of maturity of his immune system and the virulence of the pathogen. The time to overcome the threshold of the compensatory response is most likely determined (coincides) with the end of the next reproduction cycle of the pathogen, manifested by an increase in structural and functional cellular changes of a negative nature and the appearance of the pathogen in the blood and in exudates on the surface of the mucous membranes and skin. The amount of peroxides increases in cells, and eicosanoids in plasma. As the pathogen disseminates, the severity of local processes and the multiplicity of infection of the macroorganism progressively increase, accompanied by the appearance and development of vague symptoms of the disease in the infected person, creating a general feeling of discomfort. During this initial period of the disease, in addition to quantitative changes in mediators and cytokines, an imbalance in the blood coagulation-anticoagulation system begins to appear, and the patient’s condition begins to sharply deteriorate, which is usually felt by him as the onset of the disease. From this time and at the height of the disease, dramatic changes occur in the patient’s tissues and blood. The outflow of plasma into the intercellular spaces is decompensated, perivascular leukocyte infiltration, erythrocyte diapedesis and blood thickening increase, and “sludge” symptoms are formed.
Increased platelet aggregation and migration of leukocytes-neutrophils into the walls of blood vessels and perivascular clefts activate Hageman factor (factor XII), the “trigger” component of the coagulation cascade. Against the background of thrombocytopenia and leukopenia, symptoms of hypercoagulation characteristic of stage I of DIC syndrome develop.
Further progressive structural and functional disorganization of the endothelium, germinal points of bone marrow stem cells and others (in yellow fever and Marburg fever - hepatocytes and mononuclear phagocytes) leads to an increase in capillaropathy and, accordingly, toxicosis, up to the development of infectious-toxic encephalopathy (ITE) with complete loss of consciousness and perception of the environment, i.e. a state of coma develops (synonym, ITE III degree. The level of cytokines and mediators involved in pyrogenesis (PGE3, IL-1, leukotrienes), coagulation - anticoagulation fluctuates dynamically, the synthesis of colony-stimulating factors (CSF) begins to decrease. A decrease in CSF is accompanied by suppression of the process self-renewal of pluripotent stem cells; the direction of differentiation of blood cells, in particular neutrophils, and their maturation change.
The unusual content of decay products of cells affected by the pathogen, compared to the physiological norm, and the increased synthesis and secretion of eicosanoids at the initial stage of the infectious process are accompanied by an increasing insufficiency of the excretory function of the kidneys and the neutralizing activity of the liver due to ischemia, and in some hemorrhagic infections, due to its direct damage by the pathogen, occurring synchronously with the involvement of the endothelium and other cells. For HFRS, which has a slower course compared to Marburg and Ebola fevers, vascular thrombi and endothelial cell necrosis are uncharacteristic, and the clinical picture is caused by generalized capillary dilatation, edema, and infiltration of large organs and lymph nodes by granulocytes. For this nosoform, the most noticeable is the increased permeability of capillaries, including nephrons, reflecting endothelial dysfunction in combination with an imbalance of mediators and other regulatory components of hemostasis.
At the final stage of viral hemorrhagic fevers with an unfavorable outcome, thrombocytopenia persists due to the loss of platelets into aggregates and microthrombi, deficiency of Hageman factor, thrombooxane A2, and other mediators; the hypocoagulation phase smoothly passes into a state of deep hypocoagulation with the development of corresponding symptoms - the blood clotting time and platelet level increase many times over and leukocytes are as low as possible, bleeding reaches a maximum, while body temperature decreases.
Thus, the pathogenesis of hemorrhagic conditions during hemorrhagic fevers reflects a complex of dynamically occurring coupled processes, layered on top of each other, the basis of which is: - deregulation of the normal physiological ratio of the components of systems providing hemostasis (see above), due to changes in the secretory activity of cells involved to hemostasis; - anatomical and morphological exposure of the walls of blood vessels in the most active (in terms of its functional activity) part, namely in areas of microcirculation (arteriole-capillary-venule); - increased vascular permeability due to a violation of their integrity and changing levels of vasoactive components in the blood (platelets, eicosanoids, kinins, adrenaline and other pharmacologically active compounds); — lag of immunologically important protective reactions of the macroorganism, including in the form of the formation of specific antibodies, from the increase in destructive changes during the generalization of the infectious process; — the overall formation of disseminated intravascular coagulation symptoms of varying degrees of severity.
In this case, changes in the blood consistently develop through the hypercoagulation phase into the hypocoagulation stage with obligatory thrombosis and leukopenia of consumption and a shift in the neutrophil formula to the left. The rate of development of symptoms and the outcome of the disease are determined by the degree of pathogenicity of the pathogen, the constitutional characteristics and immune status of the patient.
Etiology
The causative agents of the pathology are viruses of various families that are tropic to the endothelial cells of blood vessels. They are carried by insects - ticks and mosquitoes. In the human body and some animals, microbes persist for a long time. The natural hosts of dangerous viruses are bats, squirrels, rodents, porcupines and primates.
The vector-borne route of spread of infection is the main one and is realized through insect bites.
Other ways of infection include:
- Airborne dust,
- Parenteral,
- Nutritional,
- Water,
- Contact.
Microbes enter the human body through small scratches, abrasions and wounds on the skin. The airborne dust pathway is realized by inhaling dust containing particles of animal excrement. Infection is also possible by eating contaminated foods. Health care workers caring for sick people during outbreaks are at high risk of infection.
The main pathogenetic links of hemorrhagic fever:
- Inflammation of the endothelial layer of blood vessels,
- Destruction of the vascular wall,
- Capillarotoxicosis,
- Hemorrhages in the skin and mucous membranes,
- Release of inflammatory mediators and substances with cytotoxic effects into the blood,
- Pronounced trophic changes in cells,
- Disseminated intravascular coagulation,
- Insufficient oxygen supply to tissues,
- Dysfunction of internal organs and systems,
- Massive blood loss.
Hemorrhagic fever is a dangerous pathology, which, even with timely and correct treatment, can result in the death of the patient.
HFRS - epidemiology of the disease
Mouse-like rodents are considered the main source of viral particles.
Rodents carry this infection asymptomatically. The virus is released into the environment through feces and urine.
Human infection occurs through aspiration and contact.
Attention. In aspiration infection, the virus is aspirated (inhaled) from the remains of dried rodent feces.
Contact infection occurs through direct contact with rodents, as well as materials and surfaces contaminated with their feces (hay, grain, straw, etc.).
Nutritional infection is also possible, which occurs when consuming thermally unprocessed foods contaminated with the feces of sick rodents.
For reference. Infected people do not pose an epidemic danger, since the disease is not transmitted from person to person.
The rate of natural sensitivity to the pathogen is high in all age groups.
Most often (in approximately eighty to ninety percent of all cases), HFRS is registered in males from sixteen to fifty years of age.
The disease develops mainly in rural industry workers, tractor drivers, etc.
For reference. After an infection, lifelong type-specific immunity is formed.
Natural foci of this infection are recorded throughout the world. In Russia, the disease is registered in all regions. This infection is most common in Tatarstan, Bashkortostan, etc.
There is a pronounced seasonality in the morbidity structure. HFRS is recorded mainly from May to December.
Diagnostics
Diagnosis of pathology begins with interviewing the patient and collecting anamnesis. Experts find out in which region the patient lives, whether he had contact with animals, whether he was bitten by insects. Doctors should be alert to high body temperature and hemorrhagic manifestations.
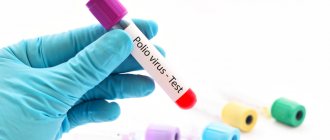
Laboratory diagnostics allows you to confirm or refute the suspected diagnosis. The most informative laboratory techniques:
- General clinical analysis of blood and urine,
- Biochemical blood test,
- Coagulogram,
- Examination of feces for occult blood,
- Serodiagnosis - RSK, RNIF, RN, RTGA, RIA,
- Immunological blood test,
- ELISA,
- PCR diagnostics,
- Isolation and study of viruses.
Etiological factors of the disease
HFRS is one of the focal natural infectious pathologies common in Eurasia. Most often, the disease is registered in China and the Russian Federation.
The causative agent of the disease is arboviruses belonging to the Bunyaviridae family. This type of virus is classified as hantavirus.
For reference. HFRS is caused by four serovars (a group of microorganisms belonging to the same species) of this virus: Puumala, Dobrava-Belgrade, Seoul and Hantaan.
Manifestations of the infectious process do not depend on the type of serovar of the pathogen.
HFRS caused by viruses such as Dobrava-Belgrade and Puumala is most often recorded in Russia.
For reference. Viruses that cause this type of HF are distinguished by a high level of sensitivity to the effects of disinfectants. solutions and ultraviolet radiation. Also, viruses are inactivated within half an hour when exposed to temperatures above fifty degrees.
At temperatures up to twenty degrees and at temperatures below -20 degrees, the virus is relatively stable. In blood samples at temperatures up to 4 degrees, the virus can persist for four days.
Causes
The disease can be caused by:
- bunyaviruses;
- togaviruses;
- arenaviruses;
- filoviruses;
- flaviviruses.
This group of diseases is characterized by natural focality. This suggests that they are most common in places where many animals live, as well as virus carriers, which include mosquitoes and ticks. Animal carriers:
- rodents (mouse fever);
- proteins;
- monkey;
- the bats.
But it is worth noting the fact that diseases are characterized by the following distribution routes: parenteral, airborne, dusty, waterborne, and foodborne.
HFRS - what is it?
For reference. HFRS is an acute viral zoonotic focal natural infection.
Characterized by:
- systemic damage to microcirculatory vascular structures,
- development of hemorrhagic diathesis,
- severe hemodynamic disorders,
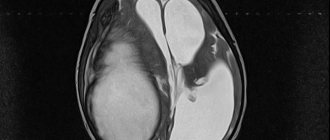
- damage to kidney tissue,
- development of severe renal dysfunction.
This type of hemorrhagic fever is also called:
- hemorrhagic nephrosonephritis,
- epid. nephrosonephritis,
- Manchurian fever, etc.
According to the ICD10 classification, this infection is classified as A98.5.
Treatment
Hemorrhagic fever requires urgent hospitalization. Treatment is carried out in the infectious disease ward of a hospital or in the intensive care unit. Patients are given hemostatic, replacement, desensitizing and symptomatic therapy. If patients are admitted to the hospital in a state of shock, they are administered Dopamine, cardiac glycosides, and Reopoliglucin.
Patients are prescribed strict bed rest and diet therapy. At the height of the disease, parenteral nutrition is prescribed, and during the recovery period - vegetable and dairy light foods enriched with vitamins PP, C, B or K. Vegetable and fruit decoctions, juices, infusions, and fruit drinks are most useful.
- Replacement therapy is an important aspect of treatment. Patients are advised to receive transfusions of blood components - intravenous administration of platelets, coagulation factors, and iron supplements.
- Detoxification therapy - intravenous administration of saline solutions, glucose solution, Hemodez, Reopoliglyukin.
- Antiviral therapy – “Amiksin”, “Ingavirin”, “Anaferon”, “Kagocel”.
- Vitamin therapy – vitamins C, R.
- Hemostatic therapy – “Vikasol”, “Ditsinon”, “Etamzilat”.
- Antihistamines - “Suprastin”, “Diazolin”, “Tavegil”.
- Antipyretics – “Ibuklin”, “Nurofen”.
- Anti-pain medications – “Ketorol”, “Pentalgin”, “Spazgan”.
- Disaggregants – “Aspirin”, “Cardiomagnyl”, “Thromboass”.
- To improve microcirculation, Heparin, Clexane, and Fraxiparin are prescribed.
- Angioprotectors and antioxidants - Actovegin, Vinpocetine.
- Diuretics for the development of renal complications - Lasix, Furosemide.
- Hemodialysis is applicable if kidney damage is observed.
Properly carried out comprehensive treatment can significantly reduce the risk of complications and speed up the recovery process.